Journal Name: Journal of Biomedical Research and Reviews
Article Type: Research
Received date: 17 September, 2018
Accepted date: 19 September, 2018
Published date: 2021-03-21
Citation: Friedenson B (2018) A Genome Model to Explain Major Features of Neurodevelopmental Disorders. J Biomed Res Rev Vol: 1, Issu: 2 (36-57).
Copyright: © 2018 Friedenson B. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The purpose of this study was to understand the role of infection in the origin of chromosomal anomalies linked to neurodevelopmental disorders. In patients with neurodevelopmental disorders, DNA’s from viruses and bacteria including known teratogens were tested against chromosome anomalies known to cause the disorders. Results support a theory that parental infections disrupt elaborate multi-system gene coordination needed for neurodevelopment. Genes essential for neurons, lymphatic drainage, immunity, circulation, angiogenesis, barriers, structure, and chromatin activity were all found close together in polyfunctional clusters that were deleted in neurodevelopmental disorders. These deletions account for immune, circulatory, and structural deficits that accompany neurologic deficits. In deleted clusters, specific and repetitive human DNA matched infections and passed rigorous artifact tests. In some patients, epigenetic driver mutations were found and may be functionally equivalent to deleting a cluster or changing topologic chromatin interactions because they change access to large chromosome segments. In three families, deleted DNA sequences were associated with intellectual deficits and were not included in any database of genomic variants. These sequences were thousands of bp and unequivocally matched foreign DNAs. Analogous homologies were also found in chromosome anomalies of a recurrent neurodevelopmental disorder. Viral and bacterial DNAs that match repetitive or specific human DNA segments are thus proposed to interfere with highly active break repair during meiosis, and sometimes delete polyfunctional clusters, and disable epigenetic drivers. Mis-repaired gametes produce zygotes containing rare chromosome anomalies which cause neurologic disorders and accompanying non-neurologic signs. Neurodevelopmental disorders may be examples of assault on the human genome by foreign DNA with some infections more likely tolerated because they resemble human DNA segments. Further tests of this model await new technology. Graphical representation of the work is shown in figure 1.
Keywords
Genome, Epigenetics, Neurodevelopmental disorders, Chromosome anomalies, Retrotransposon, Chromosome rearrangement, Neurologic disease, Birth defects, Development, Infection.
Abstract
The purpose of this study was to understand the role of infection in the origin of chromosomal anomalies linked to neurodevelopmental disorders. In patients with neurodevelopmental disorders, DNA’s from viruses and bacteria including known teratogens were tested against chromosome anomalies known to cause the disorders. Results support a theory that parental infections disrupt elaborate multi-system gene coordination needed for neurodevelopment. Genes essential for neurons, lymphatic drainage, immunity, circulation, angiogenesis, barriers, structure, and chromatin activity were all found close together in polyfunctional clusters that were deleted in neurodevelopmental disorders. These deletions account for immune, circulatory, and structural deficits that accompany neurologic deficits. In deleted clusters, specific and repetitive human DNA matched infections and passed rigorous artifact tests. In some patients, epigenetic driver mutations were found and may be functionally equivalent to deleting a cluster or changing topologic chromatin interactions because they change access to large chromosome segments. In three families, deleted DNA sequences were associated with intellectual deficits and were not included in any database of genomic variants. These sequences were thousands of bp and unequivocally matched foreign DNAs. Analogous homologies were also found in chromosome anomalies of a recurrent neurodevelopmental disorder. Viral and bacterial DNAs that match repetitive or specific human DNA segments are thus proposed to interfere with highly active break repair during meiosis, and sometimes delete polyfunctional clusters, and disable epigenetic drivers. Mis-repaired gametes produce zygotes containing rare chromosome anomalies which cause neurologic disorders and accompanying non-neurologic signs. Neurodevelopmental disorders may be examples of assault on the human genome by foreign DNA with some infections more likely tolerated because they resemble human DNA segments. Further tests of this model await new technology. Graphical representation of the work is shown in figure 1.
Figure 1
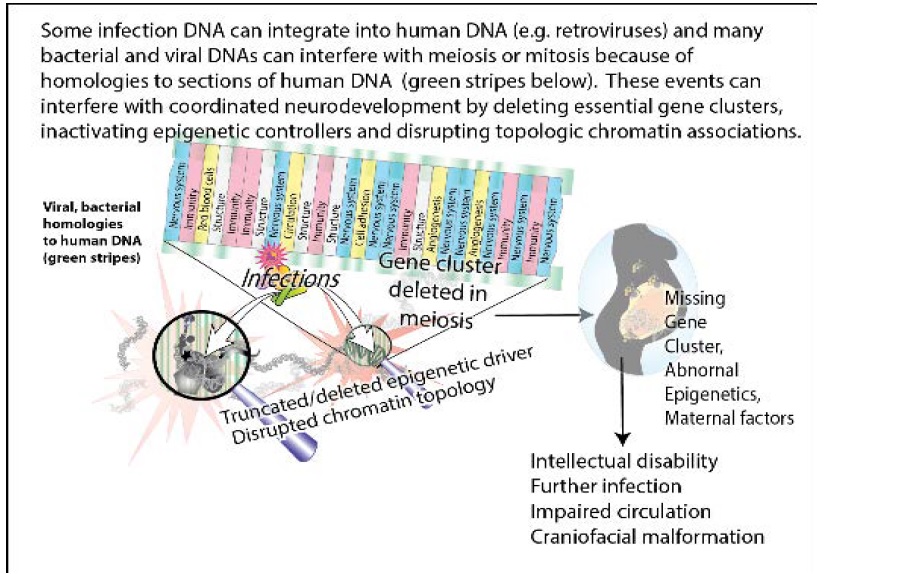
Figure 1: Graphical representation of the abstract.
Keywords
Genome, Epigenetics, Neurodevelopmental disorders, Chromosome anomalies, Retrotransposon, Chromosome rearrangement, Neurologic disease, Birth defects, Development, Infection.
Introduction
An approach to preventing neurodevelopmental disorders is to gain better understanding of how neurodevelopment is coordinated and then to identify environmental and genomic factors that disrupt it. The development of the nervous system requires tight regulation and coordination of multiple functions essential to protect and nourish neurons. As the nervous system develops, the immune system, the circulatory system, cranial and skeletal systems must be synchronized and coordinated. Neurodevelopmental disorders follow the disruption of this coordination.
A significant advance in genome sequence level resolution of balanced cytogenetic abnormalities greatly improves the ability to document changes in regulation and dosage for genes critical to the function of the neurologic system. Based on DNA sequence analyses, some chromosome rearrangements have been identified as causing individual congenital disorders because they disrupt genes essential for normal development [1-3]. There is poor understanding and no effective treatment for many of these overwhelming abnormalities. Signs and symptoms include autism, microcephaly, macrocephaly, behavioral problems, intellectual disability, tantrums, seizures, respiratory problems, spasticity, heart problems, hearing loss, and hallucinations [1]. Because the abnormalities do not correlate well with the outcome, genetic counseling is difficult and uncertain [3].
Several maternal factors are known to interfere with neurodevelopment including infection, and other lifestyle factors that subsequently alter the epigenome, but the genetic events that lead to most neurodevelopmental disorders are not understood [4]. In congenital neurologic disease, inheritance is usually autosomal dominant and the same chromosomal abnormalities occur in every cell. The present work explains how DNA from infections can cause chromosomal deletions, rearrangements, damaged epigenetic controllers and abnormal chromatin topology. Some disorders may arise when infections replicate within the CNS by taking advantage of immune deficiencies such as those traced back to deficient microRNA production [5]. Disseminated infections may interfere with the highly active DNA break repair process required during meiosis, to generate gametes with mis-repaired DNA. In the zygote, this causes chromosome anomalies. The generation of gametes by meiosis is the most active period of recombination. Double strand breaks initiate meiotic recombination and hundreds of double strand breaks occur [6]. The timing and duration of recombination during meiosis differ markedly between males and females. Meiotic recombination in sperm cells occurs continuously after puberty. Errors in spermatogenesis underlie a prevalent and recurrent gene rearrangement that causes Emanuel syndrome. The phenotype of Emanuel syndrome includes intellectual disability, and dysmorphism [2]. In contrast, recombination in ova occurs in fetal life and then meiosis is arrested until puberty. [7].
The exact DNA sequences of known pathogenic rearrangements in unique, familial and recurrent congenital disorders [1-3] makes it possible to test for a mechanism involving microbial DNA at homologous human sequences. For retroviruses, insertion hotspots can significantly change the 3D structure of the genome and disrupt essential chromatin interactions that appear to be long range in two dimensions [8]. Even rare and unique developmental disorders can be screened for homology to infections in the context of altered chromatin structure affecting the immune system, the circulatory system, the formation of the braincirculation barriers and bone development. This screening may assist counseling, diagnosis, prevention, and early intervention.
The results showed that DNAs in some congenital neurodevelopmental disorders closely matches DNA in multiple infections that extend over long linear stretches of human DNA and often involve repetitive human DNA sequences. Neurodevelopmental disorders are proposed to begin when parental infections cause insertions or interfere with meiosis at both repetitive and unique human DNA sequences. The affected sequences are shown to exist as linear clusters of genes closely spaced in two dimensions. Interference from infection can delete or damage human gene clusters and epigenetic regulators that coordinate neurodevelopment. This genomic interference accounts for immune, circulatory, and structural deficits that accompany neurologic deficits. Congenital neurodevelopmental disorders are thus viewed as resulting from an assault on human DNA by microorganisms and an example of the selection of infecting microorganisms based on their similarity to host DNA. However, a linear 2D model is sometimes inadequate to explain certain results and it is important to remember that effects in two dimensions may alter 3D topology as well [8].
Testing and verifying predictions from a viable model may however spur the development of methods for identifying contributions from infections in intractable rare disorders that are not now available. Convergent arguments from testing predictions based on any proposed model might lessen effects of limitations in currently available technology.
Materials and Methods
Data sources
DNA sequences from acquired congenital disorders were from published whole genome sequences at chromosome breakpoints and rearrangement sites [1-3]. Comparison to multiple databases of microbial sequences determined whether there were regions of the human genome near recurrent breakage sites that had significant homology. In many cases there was robust evidence that a particular human chromosome rearrangement was pathologic for the congenital disorder and this data was a point of focus.
Testing for microbial sequences
Hundreds of different private rearrangements in 62 patients with different acquired congenital disorders were tested for homology [9] against non-human sequences from microorganisms known to infect humans as follows: Viruses (taxid: 10239), and retroviruses including HIV- 1 (taxid:11676), and human endogenous retroviruses (Taxids:45617, 87786, 11745, 135201, 166122, 228277and 35268), bacteria (taxid:2), Mycobacteria (taxid:85007) actinobacteria (taxid:201174), chlamydias (taxid:51291).
Various analyses place Alu repeats into 8 subfamilies having consensus sequences (GenBank; accession numbers U14567 - U14574). To correct for errors caused by Alu repeat artifacts in microbial sequences, microbial sequences were independently compared by the same methods to all 8 consensus Alu sequences and to 442 individual AluY sequences.
Often two comparisons were run simultaneously, one
with humans excluded and one with humans included.
Homologies identified were then sorted according to
whether they involved teratogens, viruses or bacteria.
Because homologies represent interspecies similarities, “Discontinuous Megablast” was used and the ten strongest
homologies were measured in each run. Known teratogens
included, HIV-1, N. gonorrhoeae, and N. meningitidis.
Significant homology (indicated by the homology score)
occurs when microbial and human DNA sequences have more
similarity than would be expected by chance (E values). To
minimize the chances for false positives, a cutoff for E values
Chromosome localizations
When homologies were identified, the position in the target chromosomes were determined by entering the viral FASTA sequence into BLAT or into BLAST to determine its human genome location. Inserted sequences were also compared to Mus musculus GRCM38.p4 [GCF_000001635.24] chromosomes plus unplaced and unlocalized scaffolds (reference assembly in Annotation Release 106). The reference assembly set of RefSeq genomic top-level sequences (chromosomes, unplaced and unlocalized scaffolds) in a specific annotation run. Comparisons were also made to cDNAs based on 107,186 Reference Sequence (RefSeq) RNAs derived from the genome sequence with varying levels of transcript or protein homology support. Tests for contamination by vector sequences in these nontemplated sequences were also carried out with the BLAST program. Genes present in deleted segments were based on current genes identified in the UCSC Genome browser. Epigenomic modifications in associated histones in 9 model cell lines were also identified in the UCSC genome browser. In some cases, homology of inserted sequences to each other was tested using the Needleman and Wunsch algorithm.
Results
Interdependent functions are clustered together on the same chromosome segment
Anatomical relationships in the nervous system show a close relationship and synchronization with structures essential for the development of immunity, circulation and protective enclosures. In chromosome segments deleted in neurologic disorders, genes essential for the nervous system, the immune system, circulation, brain-circulatory barriers and even bone structure are located close to each other on the same linear segment of a chromosome (Figure 2). Deletions at 4q34 in patient DGAP161 and at 19q12- 13.11 in patient DGAP125 are shown in Figure 2(a) and (b), respectively as examples that are representative of the other large chromosomal deletions. The genes within each deletion belong to each of the categories tested are color coded in the figure. These clustered arrangements are crucial because their deletion has been correlated with serious neurodevelopmental disorders [1]. In many cases, and alternatively spliced forms of the same gene may be used to encode for multiple functions that must be synchronized and coordinated among diverse cells. Multiple functions in different cell types are commonly found. Hormonal signaling represents a major control mechanism [11].
Figure 2
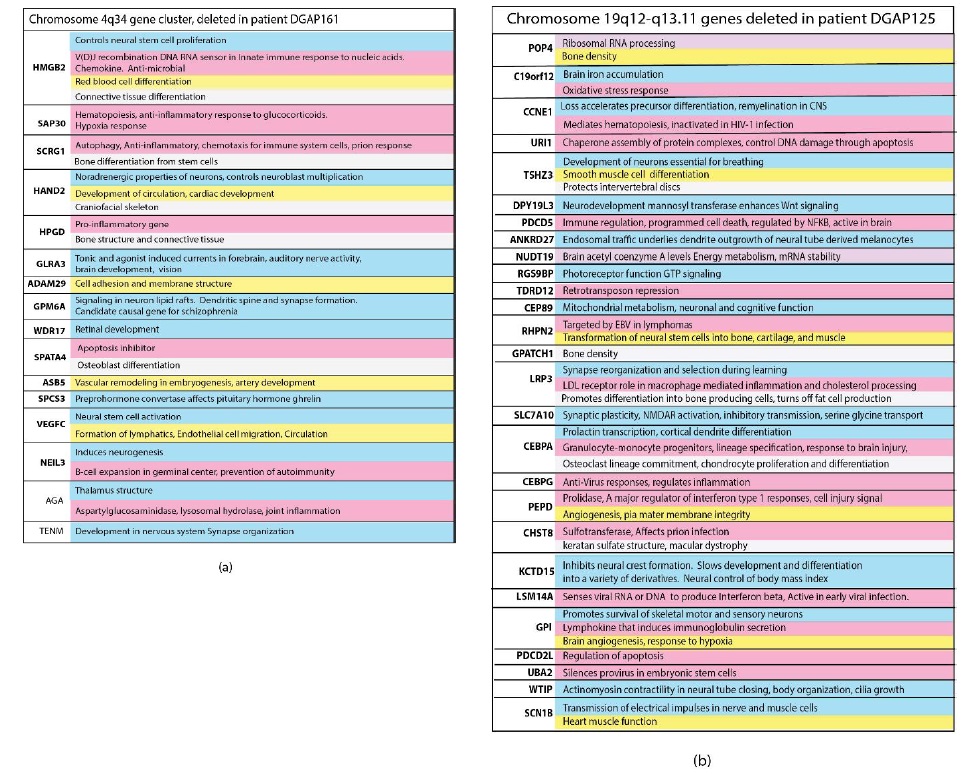
Figure 2: Close relationships between the nervous system and other essential functions developed during evolution. Genes for multiple interdependent systems appear in clusters on the same chromosomes. Nervous system genes are in close proximity to genes essential for the immune system, connections to lymphatic circulation, ability to form tight junctions, structural enclosures and chromatin control. Clusters of genes encoding these and other interdependent functions on chromosome segments are deleted in private neurodevelopmental disorders. These losses increase susceptibility to infections which are homologous to long stretches of human DNA. Genes are listed in the order they occur on the chromosome segment with the functions given to the right of the gene symbol. Blue genes are associated with the nervous system, pink with the immune system. Yellow genes have functions associated with angiogenesis, lymphangiogenesis, circulation or the development of barriers to the nervous system. Genes associated with the development of essential bone structure or connective tissues needed to protect and house the nervous system are light grey. Genes colored purple are associated with general functions required by all cells. The same gene may have several of these functions depending on its cellular location.
Many deleted gene clusters include long stretches of DNA strongly related to infections
To investigate the chromosomal segment deletions that cause neurodevelopmental disorders, homologies to infection were tested in sequences within and flanking these deleted clusters. Strong homologies to infections were found interspersed throughout. To demonstrate the extent of these relationships, the deleted 4q34 chromosome segment (Patient DGAP161 [1]) was tested for homology to the top ten homologous microbes in 200 kb chunks. Figure 3 shows that stretches of homology to microorganisms are distributed throughout chromosome 4q34. Only 10 homologous microbes are shown for each 200 kb division, but there are up to hundreds, giving a total of many thousands of potentially homologous microorganisms throughout the 4q34 deletion.
Figure 3
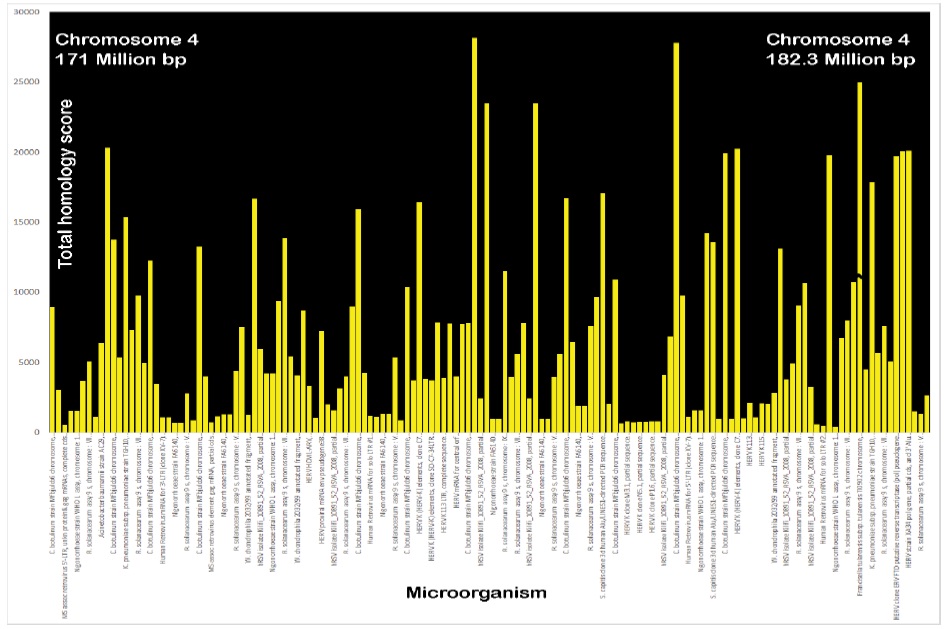
Figure 3: Example of total homologies to microbial sequences dispersed throughout 4q34 deleted chromosome segment in patient DGAP161. The top 10 homologies were tested for each 200,000 bp segment, but there are many thousands more. These homologies are listed within each of these 57 segments in order of their maximum homology scores. The Y axis plots the total homology scores and the x axis lists the individual microorganism. The mean E value was 5.3e-90 (range 0.0 to 1e-87). F. tularensis had values of 33,368 and 79,059 which were truncated at 25000 to show more detail for the other microbial matches.
A deeper analysis of the functions of genes within deleted chromosome segments predicts a predisposition to infection and correlates with symptoms of the developmental disorder. For example, deletion of chromosome 19q12-q13.11 occurs in patient DGAP125 (Figure 2). The deletion causes deficits in silencing provirus, repressing retrotransposons, responding to viruses, specifying immune cell lineages, and regulating apoptosis. The deleted 19q12-q13.11 band in DGAP125 had 76 matches to clostridium botulinum strain Mfbjuicb6. Many of these matches extended over 2800 bp at 72-73% homology with an E value of 0.0. The 5’ region included 43 of the 76 matches to clostridium covering about 2800 bp with 73% homology (E=0.0). The next million bp had another 33 matches to clostridium. There were also strong matches to Waddlia chondrophila and N. gonorrhoeae. Other shorter chromosomal anomalies are present in patient DGAP125. Stealth and HIV-1 teratogens and multiple other infections are homologous to these shorter anomalies (Figure 4).
Figure 4
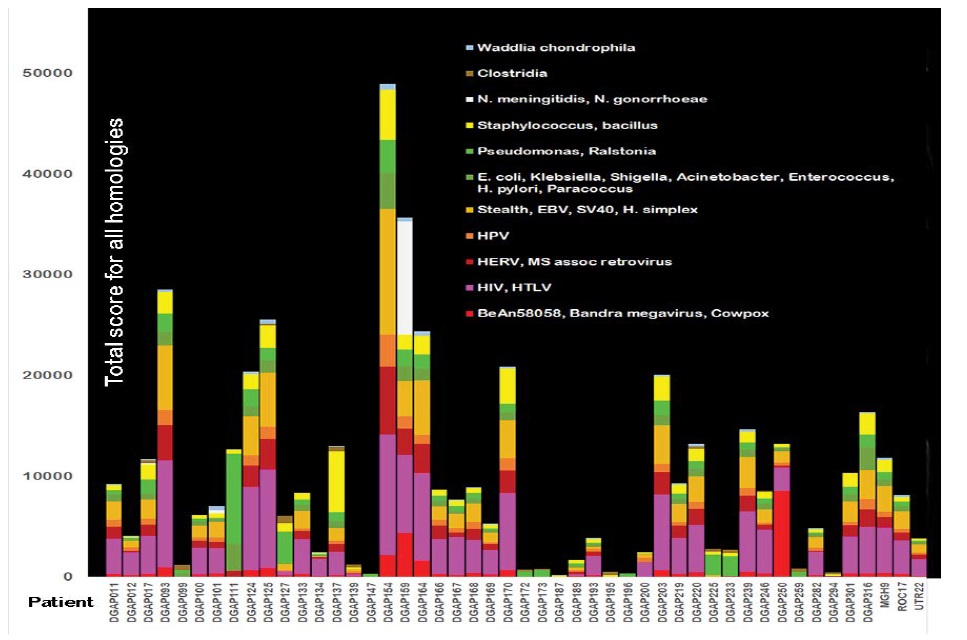
Figure 4: Total scores for homologies among microorganisms and human chromosomal anomalies in 48 patients with neurodevelopmental disorders. The contributions from all homology scores for groupings or individual microorganisms that match the chromosomal anomaly in each patient are added together to form each bar in the graph. The microorganism homology scores are color-coded according to the type of individual microorganism. Of 1986 E values, the mean was 7.3e-13 [Range 8e-11 to 2E-168]. Some of the microorganisms found are not normally pathogenic but can become opportunistic pathogens if the immune system is impaired. Endogenous retroviral sequences may interfere with recombination or repair within a single cell. In a few cases homologies were so large that they would obscure all the other data so these were limited to a score of 12,000. This was done for DGAP154 (24,873 FOR HIV/HTLV) and is indicated on the graph.
The large deletion in 19q12-q13.11 makes patient DGAP125 especially susceptible to infections and the results are potentially devastating. The damage to defenses against infection associates with coordinated genes for control of breathing, synapse organization and plasticity, neural crest formation, and transmission of nerve impulses. There are simultaneous deficits in development of brain circulation and production of connective tissue and bone. Other chromosome deletions with clustered genes have deficits in neuronal accessory functions like those in DGAP125.
Microbial DNA homologies in areas around a mutated epigenetic driver gene
In some patients with neurodevelopmental disorders, robust evidence exists that the disease is caused by a truncated driver gene [1]. The gene changes identified as underlying phenotypic drivers of congenital neurologic diseases [1] were tested against existing evidence of their ability to act as epigenetic regulators and effectors of functions like those found in the chromosomal deletions. Table 1 shows that most of the pathogenic driver genes are epigenetic regulators. For example, gene products from FOXA1, PH21A, CHD7, and TCF4 mediate or regulate chromatin remodeling; ZBTB20 and GATA3 help regulate chromatin architecture; CDKL5 inactivates a histone deacetylase, WAC regulates histone ubiquitination; SOX5 encodes a histone demethylase; EHMT1 encodes a histone methyltransferase; MEF12C is associated with histone hypermethylation in schizophrenia; KDM6A encodes an epigenetic switch that specifies stem cell fate; EFTUD2 gene product associates with chromatin binding proteins as part of the spliceosome; SNRPN is required for epigenetic imprinting; TET1 is a methylation eraser. Short DNA sequences (less than 20,000 bp) deleted from families [3] are also likely to damage epigenetic regulators. Mutations also occurred in genes within topology associated chromatin regions and in insulators (e.g. CTCF, cohesin complexes).
Table 1: Relationships of nervous system genes to epigenetic functions and to genes in the immune system, the circulatory system and structure in neurodevelopmental disorders.
| Patient | Proposed primary phenotypic drivers of anomaly /Other likely contributors d=deletion, T=truncation [1] or gene disrupted by structural variant [3] Bold entries are related to epigenetic regulation or chromatin modificatiom | Association of pathogenic neurologic mutation [24] with nervous system function | Relationship to immunity, infection | Relationship to circulation and blood brain barrier | Relationship to bone, structural requirements |
| DGAP002 | 14q12-q21.1 (d), NFKBIA (d), NKX2-1 (d) BAZ1A (d) FOXA1 (d), many other genes deleted FoxA1 mediates chromatin remodeling | NOVA1 regulates Dcc alternative splicing during neuronal migration and axon guidance in the spinal cord [25]. AKAP6 associated with cognition. FoxA1 dopamine synthesis | Fox G1 deletion in chromosome 14q12. Fox G1 regulates thymic epithelial cells [26], NFKB1A regulates immune and inflammatory responses. Deregulated in inflammatory and infectious diseases. NKX2 is needed for spleen development. BAZ1A: recovery from DNA damage. FoxA1 is characteristic of a population of regulatory T-cells. Downregulated in parasite infections. | FoxA1 can direct lineage specific differentiation into blood cells, neurons, or myocytes. | FoxA1 regulates bone formation through bone morphogenetic protein, required for intervertebral disks |
| DGAP011 | FGFR1 (T), FGF regulates chromatin organization during development | Axon guidance [27] | Required for proinflammatory cytokine expression [28] FGFR1 may contribute to interaction between NK and T-cells. | Helps protect and maintain the blood brain barrier [29] Associated with vascular remodeling. | Skeletal and craniofacial development, chondrogenesis and osteogenesis |
| DGAP012 | PHF21A (T), chromatin regulator disrupted at breakpoint | Encodes BHC80, a component of a BRAF35 histone deacetylase complex. Represses neuronal-specific genes, of fundamental importance in the development of both neuronal and nonneuronal tissues [30]. | Histone deacetylase interacts with repressor element 1 that controls the transition of neuronal cells to glial cells through nitric oxide free radicals [31]. | Promotes angiogenesis, endothelial cell survival. Histone deacetylase protects against ischemia and reperfusion injury. | Not found |
| DGAP093 | CDKL5 (T) disrupted at breakpoint, phosphorylates HDAC4 histone deacetylase causing cytoplasmic retention [32]. | CDKL5 Rett syndrome, a post-natal neurologic disorder, appearing after a period of near normal development. Mediates MECP2 phosphorylation Induced by methylation inhibitors. Seizures may reflect CDKL5 role in dendritic arborization. | Deregulated inflammatory response in microglia [33]. | PI3K-Akt pathway enriched (may be involved in tight junction opening) Affects red blood cell shape. | Not found |
| DGAP096 | WAC (T), at breakpoint, disrupted by translocation regulates histone ubiquitination | Required for cognition, prevention of intellectual disability, behavior disorders. | Required for autophagy and required for autophagosome formation [34]. DNA damage checkpoint activation. | Required for normal hematologic function. | Not found |
| DGAP099 | ZBTB20 (T), binds to genes that control chromatin architecture including MEF2c and SAT2b | Brain corpus callosum development. Binds to genes controlling neuronal subtypes | Plasma cell differentiation, impaired innate immunity dependent on TLR2 and TLR4 [35]. | Promotes generation of astrocytes involved in blood-brain barrier [36]. Controls expression of VEGFA in some settings. | Regulates chondrocyte differentiation, |
| DGAP100 | KDM6A (T). Histone Demethylase and Epigenetic switch to specify stem cell fate [37] | Maturation of functional synapses in cerebellar cortex, regulation by BDNF. Epigenetic switch to regulate stem cell lineage fate | Demethylase KDM6a epigenetically promotes IL-6 and IFN-β production in macrophages | Stem cell migration of blood cell precursors [38] | Promotes osteogenic differentiation |
| DGAP112 | 12p12.1-p11.22 (d), SOX5 (d), KRAS (d); MED21(d) SOX5 is a histone demethylase at breakpoint disrupted by rearrangement. MED21 at breakpoint may be required to displace histones to enable heat-shock response [39]. | SOX5: Specifies cell fate of dorsal interneurons. | SOX5: Th17 cell differentiation; Linked to cytoplasmic engulfment protein ELMO1 [40] LRMP, lymphoid restricted membrane protein High levels found in germinal B-cells; KRAS involved in an innate immune pathway for recognizing DNA damage [41] | SOX5 interacts with tight junction proteins such as ZO-5, occluding and claudin [42]. May control differentiation of neural stem cells into astrocytes. KRAS may orchestrate circulation and blood vessel formation. | SOX5 Marker gene for cartilage formation |
| DGAP124 | NRXN1 (T) Repressive histone marker at breakpoint disrupted by rearrangement, controls choice of NRXN splicing isoform. Repressed by histone methyl-transferase [43] | Surface recognition molecule critical for organizing and forming synapses; thousands of isoforms due to alternative splicing. Some forms interact with C1qls in synaptic cleft suggesting relation to pruning connections [44]. | C1qls belongs to the inflammatory mediator TNF superfamily involved in innate immunity. Peptide derived from NRXN blocks formation of staphylococcal biofilms [45] | NRXN1 Regulates signaling for hematopoiesis [46]. | Not found |
| DGAP125 | 19q12-q13.11 (d) 19:28,105,514-34,809,581 | See Fig 2 | See Figure2 | See Figure2 | See Figure2 |
| DGAP127 | PAK3 (T), compensates for PAK1 which interacts with histone H3 | Dendritic cell growth. Synaptic plasticity. | Markedly reduced PAK3 expression occurs in prion diseases such as Creutzfeldt-Jacob disease in humans and scrapie in animals [47] MAPK signals, induced by activation of T-cells. P21 activated kinase. | Actin organization and migration, spine morphogenesis | Not found |
| DGAP133 | 6q13-q14.1 (d) | See Figure 2; SMAP1 defective neurite outgrowth; FILIP1 developmental signaling; LCA5 blindness; SENP6 | MYO6: endocytosis in macrophages. Inability to process pathogens, defects in autophagy; IRAK1BP1 deregulated. Autophagy. | Structural deficits in basement membrane as part of blood-brain barrier, deletion includes collagen genes 19A, 12A | Col9A connective tissue, bone strength (Figure2) |
| DGAP139 | 13q14.2 (d) See Figure 2. | Intellectual disability associated with amyloid deposition due to RB1, and ITM2B losses. PHF11 DNA double strand break resection, homologous recombination [48]. | Contains genes essential for immunity: leukotriene receptor, TRIM13 deleted in lymphocytic leukemia; RB1 potential guardian of immune system; NUDT15, removal of oxidative damage; | LPAR6 cell-cell contact sites in endothelial cells. Proper vasculature formation. DLEU2 control of vascular endothelial cells. | DLeu2: Bone mineral density |
| DGAP142 | MBD5 (T) Epigenetic regulator at breakpoint Histone acetylation disrupted by rearrangement. | Associated with Angelman syndrome and 2q21.3 microdeletion. Contributes to a spectrum of neurodevelopmental disorders, including autism and schizophrenia, Insufficiency causes behavioral problems, cognition and motor delay. | Severe infections in the first year of life [49]. | Iron metabolism Control of ferritin levels | Not found |
| DGAP145 | EFTUD2 (T) [RNA splicing regulator in spliceosome] at breakpoint disrupted by rearrangement Connects methylated histone to regulated RNA splicing [50] | In model system, mutation increases apoptosis and mitosis of neural progenitors [51] Essential for splicing, | Innate immunity regulator | Pulmonary venous drainage, normal heart function | bone development, mutation associated with microcephaly |
| DGAP147 | NALCN (T) Disrupted at breakpoint | Leak conductance in neurons essential for function. | Highly adaptable orphan pore ion channel required for respiration, osmoregulation, scaffold for Src kinases. Src kinases are required to defend against infection | Not found | Not found |
| DGAP154 | Xq25 (Duplication) | XIAP: synaptic depression and axon regeneration, long term learning. THOC2 increases neurite extension. | XIAP: x-linked inhibitor of apoptosis; Overexpression of XIAP suppresses apoptosis, inhibits caspases and favors virus infection [52] | SMAD5: Gap junction and tight junction formation; vascular properties; hematopoietic stem cell development | SMAD5: Bone osteogenesis |
| DGAP155 | EHMT1 (T)/GLP: Encodes a histone methyltransferase, an epigenetic regulator. | EHMT1 Impairs expression of protocadherins, regulators of neuronal diversity, growth and development [53]. Deficiency causes increased hippocampal cell proliferation [54]. Required for memory, learning. and to repress non-neuronal genes in the brain | Establish expression potential for IFN genes. Required for optimal cytokine response to infection [55] Requirement for memory formation and dysregulated cytokine response predicts learning disability. | Protects against cardiac hypertrophy [56], congenital heart disease | Osteogenesis |
| DGAP157 | FOXP1 (T)”PRMT5 recruitment to the FOXP1 promoter facilitates H3R2me2s, SET1 recruitment, H3K4me3, and gene expression” [57] Disrupted by rearrangement. | Controls gene expression required for spatial learning and synaptic plasticity [58] | T-cell regulation, cooperates with NFKB for survival of B-cells | Induces collagen synthesis [59] | Essential for craniofacial development [60] |
| DGAP159 | 10p15.3-p14 (d) GATA3 (d) See Figure 2. GATA3 controls enhancer accessibility, recruits chromatin remodeler to reprogram cells. | GATA3 development of neurons that contact cerebrospinal fluid | GATA3 is an immune regulator. Loss increases, Susceptibility to infection | Cell adhesion and cell junctions. | Embryonic articular cartilage differentiation. |
| DGAP161 | 4q34 (d) (See Figure 2) | AGA deletion causes lysosomal storage disease with brain damage. SPCS3 Pituitary hormone production. TENM nervous system development | HMGB2 encodes an RNA/DNA sensor in innate immune response to nucleic acids HPGD a proinflammatory gene; VEGFc lymphatic development and trafficking; artery development and vascular remodeling. ADAM29 cell adhesion and membrane structure. | Red blood cell differentiation, circulation, cell adhesion, vascular remodeling | Craniofacial skeleton, bone structure, bone differentiation, osteoblast differentiation |
| DGAP164 | NODAL (T) targets kdm6b (an epigenetic regulator of neuronal plasticity) signaling in mouse brain. Rearrangement also disrupts TET1 (a methylation eraser (demethylase) | NODAL Controls neuron survival by inducing neuroprotective activity. Regulates early embryonic development. | Nodal=”activin a” which encodes a mediator for NK dendritic cell functions. Spleen, a part of the immune system, may not develop correctly, | Angiogenesis, red blood cell formation | osteoclast formation, chondrogenesis |
| DGAP166 | SCN1A (T) | Brain morphology, memory [61] Sodium ion permeability of excitable membranes. Sensory perception of pain. | Mutation may predispose to viral infections that cause acute encephalopathy. | Not found | Not found |
| DGAP169 | NR2F1 (T) Disrupted by rearrangement, Induces global chromatin repression | Controls identity of cortical neuron progenitors. Induction of GABA positive neurons, | Not found | Angiogenesis, lymphomagenesis [62] | Patterning of the upper jaw [63] |
| DGAP173 | 11p14.2 (d) | ANO3: cervical dystonia; BBOX1 schizophrenia | Mucin15, is essential for mucin barriers to infection, | Not found | Not found |
| DGAP186 | NR5A1 (T) disrupted by rearrangement, participate in resetting pluripotency | Affects hypothalamus circuit. Sex development and formation of steroidogenic tissues. | Downregulated by influenza infection, activated by T-cell stimulation Autoimmunity | Regulator of angiogenesis [64] | Not found |
| DGAP189 | SOX5 is a histone demethylase | Specifies cell fate of dorsal interneurons. | T-cell differentiation | Sox5 interacts with tight junction proteins such as ZO-5, occluding and claudin [42]. May control neural stem cell differentiation into astrocytes. | Marker gene for cartilage formation, chondrocyte differentiation |
| DGAP190 | SMS (T) Drives interactions among nucleosomes. | Spermine synthetase gene. Polyamines such as spermine are essential for growth and differentiation. Spermine controls ion channel activity and has a role in cognitive function [65]. | Spermine synthesis impairs biofilm formation by n. gonorrhoeae. Spermine may be used as a nitric oxide releasing carrier | Regulates openings of lymph pores and regulates absorption [66] | Osteoblasto-genesis, bone phenotype |
| DGAP193 | SPAST (T) | Mutation causes hereditary spastic paraplegia. Critical for complex microtubule arrays in axons, spindles and cilia. Gene controls endosome tubule fission, mannose-phosphate endosome-Golgi trafficking and lysosome structure | Oxidative stress associated with inflammation [in prostate cancer] | Regional blood flow to cerebrum | Inhibits bone morphogenic protein signaling |
| DGAP199 | NOTCH2 (T) disrupted at breakpoint by rearrangement. | Pathway is essential for differentiation and survival of neurons, axon development | Intestinal immunity in dendritic cells, mucosal immunity, IFN secretion by T-cells, promotes T-cell cytotoxicity | Arterial differentiation and function, uterine blood vessel remodeling | Osteoblast function to regulate bone formation |
| DGAP201 | AUTS2 (T) Disrupted at breakpoint by rearrangement. Associated with chromatin structure | Associates with brain development [67], controls polycomb repressive complex1 Regulates actin cytoskeleton to control migration and projections from neurons [68]. | A component of the polycomb complex guides T-cell differentiation, maintains genes targeted by EBV. Regulates developmental gene MSX1 in NK T-cells and in lymphoid progenitors [69]. Regulates Rho GTPases [70], essential to protect from enteropathogenic bacteria [71] | Not found | Not found |
| DGAP202 | KDM6A (T) Histone di/tri demethylase disrupted at breakpoint by rearrangement. | Maturation of functional synapses in cerebellar cortex, regulation by BDNF | KDM6a epigenetically promotes IL-6 and IFN-β production in macrophages | Stem cell migration of blood cell precursors [38] | Promotes osteogenic differentiation |
| DGAP211 | SATB2(T) Disrupted at breakpoint by rearrangement. Epigenetic functions, interacts with chromatin remodelers. | Modulates neuron excitability. Helps specify neuron subtype. Characteristic of migrating glia-like cells that differentiate into integrated neurons in neonatal cortex [72] | Autophagy, senescence, Cytoskeletal organization. Enhances gene expression in pre-B cells | Not found | Up regulated during guided bone regeneration of the craniofacial region. |
| DGAP219 | CUL3 (T) Ubiquitin ligase recruited to regulate chromatin pattern of lymphoid effector programs. Disrupted at breakpoint of rearrangement. | Component in Ubiquitin ligase complex collaborates with other complexes. Regulation of this pathway controls brain size and connectivity. Pathway affected may be involved in autism and schizophrenia. | Regulates levels of reactive oxygen species, ion handling in brain | Determine surface level of integrins in endothelial cells, angiogenesis. Mediates VEGFR signaling. Epigenetic regulator | Integrates collage secretion into craniofacial bone formation [73] |
| DGAP232 | SNRPN-SNURF (T)/ SMN; PWCR; SM-D; sm-N; RT-LI; HCERN3; SNRNP-N; Required for epigenetic imprinting. | Ability of neurons to regenerate axons [74] | EBV/HHV-4 infection has potential to induce anti-Sm antibodies [75]. HIV-1 Tat associates with snRNP [76]. CMV may have a role in regulation of autoantibodies to snRNPs SNRPN imprinting center | Not found | Promotes osteogenic differentiation of bone marrow embryonic connective tissue stem cells. |
| DGAP235 | MBD5 (T) Epigenetic regulator histone acetylation disrupted at breakpoint of rearrangement. | Associated with Angelman syndrome and 2q21.3 microdeletion. Contributes to a spectrum of neurodevelopmental disorders, including autism and schizophrenia, Insufficiency causes behavioral problems, cognition and motor delay. | Severe infections in the first year of life [49]. | Iron metabolism Control of ferritin levels | Not found |
| DGAP239 | CHD7 (T) Chromatin remodeler. | Regulates adult neurogenesis [77], promotes neural stem cell proliferation | Mutation associated with severe combined immunodeficiency | Not found | Needed for craniofacial development |
| DGAP244 | CTNND2 (T) | Promotes neuronal differentiation [78] Cerebellar development, myoclonic tremor and epilepsy, anxiety, cognition. | Significantly enriched in brain inflammation in malaria. Induces NFKB inflammatory response related to infection. [79] | Not found | Spine architecture |
| DGAP278 | SNRPN-SNURF (T)/ SMN; PWCR; SM-D; sm-N; RT-LI; HCERN3; SNRNP-N; Required for epigenetic imprinting | Ability of neurons to regenerate axons [74] | EBV/HHV-4 infection has potential to induce anti-Sm antibodies [75]. HIV-1 Tat associates with snRNP [76]. CMV may have a role in regulation of autoantibodies to snRNPs SNRPN imprinting center | Not found | Promotes osteogenic differentiation of bone marrow embryonic connective tissue stem cells. |
| DGAP301 | MEF2C (T), Associated with disease histone hypermethylation [80].. Disrupted by rearrangement | Regulates cortical synapses and behaviors [81] | T. spiralis infection; skeletal pain in acute infectious mononucleosis [82]; increased expression of inflammatory cytokine IL-33 in HIV infection. Activated by inflammation. Inhibits granulocytes in favor of monocytes, T-cell receptor apoptosis [83] | Controls proliferation and migration of vascular smooth muscle cells. Targets aquaporin channels to regulate angiogenesis and vasculogenesis of endothelial cells [84]. Responds to VEGFA. | Required to initiate chondrocyte hypertrophy. Antisense transcript associated with bone mineral density |
| DGAP316 | 18p11.32-p11.22 deletion: PHF21A (T) PHF21A Encodes BHC80, a component of a BRAF35 histone deacetylase complex. | This histone deacetylase interacts with repressor element 1 that controls the transition of neuronal cells to glial cells through nitric oxide free radicals; | TGIF1 immune homeostasis, regulation of CD4+ T-cells; NDUF2: control of stress responses | Not found for PHF21a, but contained in deletion | Not found for PHF21a but contained in deletion |
| DGAP317 | 6q14.1 (d) TBX18 (d): IRAK1BP1; IBTK | See Figure2 contains LCA5, ELOV4 genes for vision, HTR1B gene for neurotransmission. DOPEY nerve myelin formation | IRAK1BP1 encodes an interleukin 1 receptor associated kinase 1 binding protein IL-1 receptor associated kinases are critical regulators of innate immunity. TBX18 is overexpressed in areas of inflammation and interstitial fibrosis; IBTK Regulates inflammation induced by NFKB pathway in macrophages and neutrophils See Figure 2. | Heart muscle function | Not found |
| MGH7 | GRIN2B (T) Reflects changes in methylation levels in epilepsy [85] | Neuronal migration and axon projections in developing brain. Long term depression of hippocampus membrane currents, synaptic plasticity | Associated with maternal herpes simplex virus 2 infection. In some rat strains may respond to prenatal stress [86] | Not found | Not found |
| MGH8 | CHD8 (T) As a chromatin insulator it blocks effects of nearby enhancers. CHD8 interacts with the insulator binding protein CTCF. CTCF-CHD8 complex insulates and epigenetic regulates at active insulator sites. | Enhanced expression in inhibitory neurons causes excess of excitable neurons [87]. Brain development | Strongly upregulated by maternal immune activation in response to infection during pregnancy. ATP dependent chromatin remodeler [88]. | Not found | Not found |
| MGH9 | TCF4 (T)Regulated by histone deacetylases. Binding sites overlap histone acetylation of enhancers [89] | Regulates synaptic plasticity and memory. Recruits histone acetyl transferases to affect chromatin remodeling [90] | Regulates susceptibility to HIV-1 in macrophages and resistance of monocytes; down regulated by viral infection; represses inflammation | Angiogenesis | Osteogenesis, Chondrogenic differentiation |
| NIJ2 | PHIP (T), linked to histone methylation [91] Disrupted at breakpoint by rearrangement. MYO6 (T) | PHIP [Pleckstrin homology domain only] NMDA receptor trafficking and synaptic plasticity. MYO6 neurotransmission, receptor endocytosis | PHIP (pleckstrin homology domain interacting protein): Pleckstrin homology domains regulate macrophage migration into inflammatory sites and differentiation; MYO6: endocytosis in macrophages. autophagy. | Potential role in angiogenesis [92] | Bone metabolism |
| NIJ5 | IL1RAPL1 (T) | Synapse formation, cognition | Upregulation increases innate and adaptive immune responses. Apoptosis | Not found | Skeletal growth |
| NIJ6 | KAT6B (T)/ MORF/ MYST4 Lysine acetyl transferase. | Corpus callosum formation, | Controls lineage differentiation in development of immune system. Involved in pathogenesis of hematologic malignancies [93] | Hematopoiesis | Skeletal formation, premature skull suture fusion |
| NIJ14 | NFIA (T) regulated by chromatin structure [94] | Brain formation, corpus callosum development, spinal cord and neocortical development | In pathway that regulates inflammation, controls myeloid cell differentiation and maturation in septic mice. | Balance between oligodendrocytes and astrocytes. | Embryonic articular cartilage differentiation |
| NIJ15, NIJ6 | MYT1L (T) | One of the transcription factors needed to convert cells to neurons | On infection induced acute drop in oxygen tension [95] | Not found | Not found |
| ROC4 | CAMTA1 (T) | Long term memory formation, ALS survival, behavior, ataxia | Controls NFAT signaling. Integrates stress responses Associated with shifts in microbiome [96] | Controls calcium signals that activate a cardiac gene program | Bone differentiation |
| ROC17 | AUTS2 (T Associated with H3K4me3 and epigenetic control | Regulates actin cytoskeleton to control migration and projections from neurons [68]. Replication hotspot where copy number variation occurs. | A component of the polycomb complex, guides T-cell differentiation, maintains genes targeted by EBV. Regulates developmental gene MSX1 in NK T-cells and in lymphoid progenitors [69]. Regulates Rho GTPases [70] which protect from entero-pathogenic bacteria [71] | Not found | Not found |
| ROC23 | TCF12 (T) | Learning and memory, early neuron proliferation during brain development [97] | Required for T-cell development in combination with E2A [98] Critical regulator of mesoderm and hematopoietic specification [99] | Controls cadherin at cell-cell junctions | Osteogenic cell differentiation, fusion of fibrous skull sutures |
| ROC43 | SOX5 is a histone demethylase | Specifies cell fate of dorsal interneurons. | T-cell differentiation | Sox5 interacts with tight junction proteins such as ZO-5, occluding and claudin [42]. May control differentiation of neural stem cells into astrocytes. | Marker gene for cartilage formation |
| ROC62 | SNRPN-SNURF (T) Required for epigenetic imprinting | Ability of neurons to regenerate axons [74] | EBV/HHV-4 infection has potential to induce anti-Sm antibodies [75]. HIV-1 Tat associates with snRNP [76]. CMV may have a role in regulation of autoantibodies to snRNPs SNRPN imprinting center | Not found | Promotes osteogenic differentiation of bone marrow embryonic connective tissue stem cells. |
| UTR7 | NFIX (T) controlled by methylation | Differentiation of stem cells in hippocampus | Critical control of early B-cell lymphopoiesis and myelopoiesis. | Survival of immature hematopoietic cells | Bone development, density |
| UTR12 | DYRK1A (T), histone phosphorylation | Dopaminergic cell survival, brain growth | DYRK1A Regulates T cells [100], controls bone homeostasis | Angiogenesis in endothelium | Bone homeostasis, chondrocyte development, skeletal formation |
| UTR13 | MBD5 (T) Epigenetic regulator disrupted in topology associated domain | Associated with Angelman syndrome and 2q21.3 microdeletion. Contributes to a spectrum of neurodevelopmental disorders, including autism and schizophrenia, Insufficiency causes behavioral problems, cognition and motor delay. | Severe infections in the first year of life [49]. Epigenetic regulator. | Iron metabolism Control of ferritin levels | Not found |
| UTR17 | ZBTB20 (T), binds to genes that control chromatin architecture including MEF2c and SAT2b) | Regulation of neurogenesis | Lymphoid development and function, Development and function of B-cells, Plasma cell differentiation, [35] | Astrocyte response to injury, gliogenesis | Terminal differentiation of hypertrophic chondrocytes |
| UTR20 | FOXP2 (T) | Increases neuronal differentiation, regulates development of neural progenitors in mammalian cortex [101] | Levels altered in response to maternal infection with Influenza virus in mice. Robust evidence links maternal infections to schizophrenia [102]. Regulatory T-cell function. | Controls cadherin expression in neuroepithelium [103] | Coordinates bone formation and development |
| UTR21 | NSD1 (T) lysine methylation | Cerebellar hypoplasia. Cytoskeletal regulation in neurons, directional cortical neuron migration | Caspase activation in macrophages. | Lymph circulation. | Bone development |
| UTR22 | 2q24.3 (d) SCN9A (d) | Several sodium channel genes required to control neuronal excitability deleted. TTC21B gene for organizing brain development; STK39 regulates brain volume. | NOSTRIN controls nitric oxide production, CERS6 related to T-cell activity and apoptosis. | FIGN affects astrocyte migration, XIRP2, CERS6 cardiac health | TTC21B craniofacial development |
| Affected member of family 1 | NCALD1 exists as myristoylated and non myristoylated forms. Histone deacetylase 11 is a myristoyl hydrolase [104]. | Calcium sensor in neurons | Endocytosis | Calcium signaling | |
| Affected member of family 1 | NPL - N-acetylneuraminate lyases regulate cellular concentrations of sialic acid by mediating the reversible conversion of sialic acid into N-acetylmannosamine and pyruvate | May lead to altered sialic acid metabolism required for brain development; e.g neuronal cell adhesion and myelin associated glycoproteins [105] | Enzyme with markedly increased expression in neutrophils and monocytes during inflammatory response after stroke [106]. | Enzyme activity demonstrated in blood | Strongrelationships between sialic acid and bone formation |
| Affected member of family 1 | BCAR3. A candidate epigenetic mediator for increased adult body mass index in socially disadvantaged females [107] | Endothelin-1 signaling. Edothelin 1 is a potent vasoconstrictor | Arrests cell cycle in response to tensile stress [108] | ||
| Affected member of family 2 | ZNF423 - zinc finger protein 423 epigenetic modifications associated with obesity [109], methylation marker for age prediction. | Cell division and repair of Purkinje neuron progenitors | Development of B-lymphoid cell lineage | Osteoblast vis adipocyte lineage [110], osteoarthritis | |
| Affected member of family 3 | RAP1GDS1 - RAP1 GTP-GDP dissociation stimulator 1[111]. Antagonizes some histones. | Orients migration of multipolar neurons during development [112], | T-cell homeostasis, limits TLR inflammatory response | Vascular endothelial junction stability | Bone growth |
Figure 4 graphically relates DNA sequences in all viruses and bacteria in the NCBI database to DNA sequences around breakage sites based on the study of Redin and colleagues [1]. In most cases, there is robust evidence that the chromosomal anomaly at breakage sites causes the neurodevelopmental disorder but there may also be cryptic rearrangements elsewhere [2,3,12]. The figure shows wide differences in the total homology scores of microbes vs humans in individual disorders. Patient DGAP154 has total homology scores of well over 50,000 while other patients (DGAP142, 172, and 173) are well below 1000.
Within a few hours of fertilization, global demethylation occurs and both maternal and paternal genomes reprogram to highly open chromatin [13]. Removal of these epigenetic marks allows a sudden increase in L1 retrotransposon expression which induces an innate immune response. Many of the chromosome anomalies studied contain L1- retrotransposon sequences. L1 transcriptional activity is controlled by DNA methylation and by piwi interacting RNA. Loss of epigenetic drivers can interfere with the function of these epigenetic control mechanisms (Table 1). Figure 5 summarizes this data and shows driver genes have epigenetic control functions and most of these functions impact chromatin 3D structure and multiple systems essential for neurodevelopment.
Figure 5
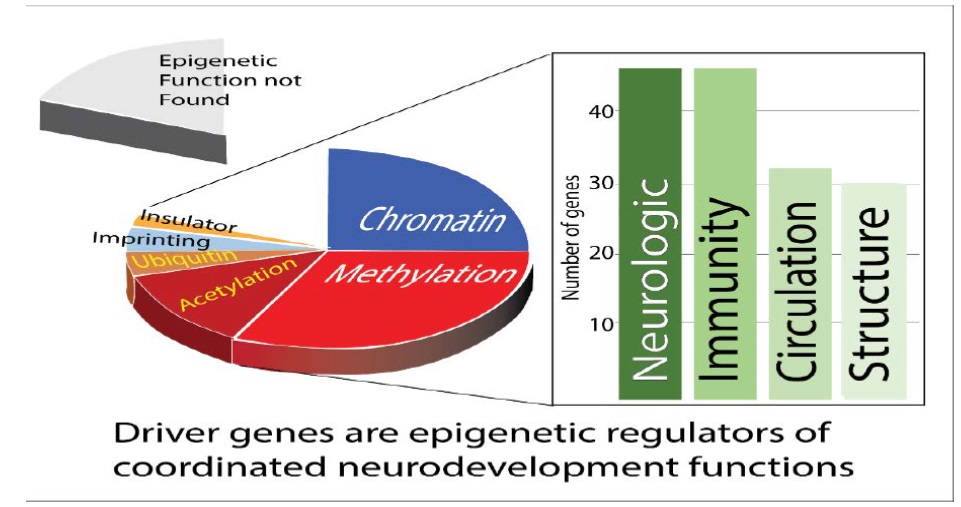
Figure 5: Total scores for homologies among microorganisms and human chromosomal anomalies in 48 patients with neurodevelopmental disorders. The contributions from all homology scores for groupings or individual microorganisms that match the chromosomal anomaly in each patient are added together to form each bar in the graph. The microorganism homology scores are color-coded according to the type of individual microorganism. Of 1986 E values, the mean was 7.3e-13 [Range 8e-11 to 2E-168]. Some of the microorganisms found are not normally pathogenic but can become opportunistic pathogens if the immune system is impaired. Endogenous retroviral sequences may interfere with recombination or repair within a single cell. In a few cases homologies were so large that they would obscure all the other data so these were limited to a score of 12,000. This was done for DGAP154 (24,873 FOR HIV/HTLV) and is indicated on the graph.
Pathogenic driver gene mutations are roughly equivalent to large chromosome deletions
Because most identified driver genes of neurodevelopmental disorders are epigenetic regulators or effectors, the functions they control were compared to those found in chromosomal deletions that cause neurodevelopmental disorders. Virtually all pathogenic driver genes or chromosomal deletions in congenital neurologic disorders have strong relationships to effects on the immune system, angiogenesis, circulation and craniofacial development. Deletions in affected members of families with neurodevelopmental disorders are also consistent with this conclusion (Table 1).
Multiple infections identified by homology match signs and symptoms of neurodevelopmental disorders
In the 48 patients shown in Figure 4, multiple infections are candidates to cause the signs and symptoms in each individual (Table 2). 8 patients have growth retardation or “short stature” which can be caused by exposure to many infections in the second or third trimester. The absence, delay, or impairment of speech were found in 20 of 48 patients [1]. Multiple infections can cause these problems. For instance, HIV-1 causes white matter lesions associated with language impairments, and impaired fetal growth. There are nearly 50 matches to HIV-1 DNA in the chromosome anomalies of 35 patients.
Table 2: Recurrent infections found to have homology to chromosomal abnormalities in neurologic birth defects can cause developmental defects.
| Recurrent Infections matching DNA rearrangements in congenital neurologic disorders | CNS, physiologic and or teratogenic effects if known. | Numbers of Patients with chromosomal abnormalities with significant homologies to this infection |
|---|---|---|
| HIV-1, HIV-2 | Impaired fetal growth, premature delivery, chorioamnionitis, deciduitis, immunodeficiency. HIV-1 causes white matter lesions associated with language impairments. HIV-1 infects CNS by targeting microglial cells. | 600 matches in 36 patients |
| HPV16 | Part of a group of infections that coexist with other viral infections, bacterial infections, and chemicals in autism syndrome disorders [113]. | 35 patients |
| Staphylococcal infection, (s. aureus, s. capitis, s. epidermidis) | May infect brain and interfere with normal nerve transmission. Causes meningitis, Brain abscess. Major hospital pathogens | 206 matches in 40 patients |
| Stealth viruses, CMV, HHV-4/EBV, herpes simplex, SV40 | Stealth viruses are mainly herpes viruses such as CMV that can avoid recognition by the human immune response. Associated with neurologic impairments, hearing loss, ophthalmic problems. | 425 matches in 39 patients |
| Ralstonia solanacearum | Opportunistic bacterial pathogen, associated with pneumonia and neonatal sepsis, especially in patients with compromised immunity on ventilator. Has some resemblance to other human pathogens including Borrelia, Bordetella, Burkholderia, and a “blood disease pathogen.” | 158 matches in 44 patients |
| HTLV-1 | Associated with neuropathy. Immune mediated disease of the nervous system affects spinal cord and peripheral nerves. Fetal neural cells are highly susceptible to infection | 24 patients |
| BeAn 58058 virus, Bandra megavirus | BeAn 58058 is almost identical (97%) to cotia virus which can infect human cells [114]. Represents a distinct branch of poxviruses [115] | 50 matches in 36 patients |
| Klebsiella pneumoniae, e coli | Neonatal pneumonia and sepsis. | 35 patients |
| Human respiratory syncytial virus Kilifi | Neonatal pneumonia, trouble breathing | 7 patients: DGAP099, DGAP127, DGAP137, DGHAP159, DGAP167, DGAP172, DGAP225 |
| Waddlia Chondrophila | Recently discovered. Causes preterm birth, miscarriage, spontaneous abortion. Chlamydia like micro-organism. Survives in macrophages, multiplies in endometrial cells | 26 different patients |
| Neisseria meningitidis, N. gonorrhoeae | Bacteria that can cause damage to human DNA. Premature birth, miscarriage, severe eye infection in infant. Antibodies to N. gonorrhoeae impair out-growth of neurites, and cross react with specific brain proteins. | Patient DGAP159 shows 25 different homologies to N. meningitidis. DGAP017 and in DGAP101 have homology to N. gonorrhoeae. |
| Pseudomonas putida | Gram-negative bacterium found in cases of acute bacterial meningitis [116]. Reported in contaminated water and heparinized flush solutions [117] | 13 patients: DGAP012, DGAP93, DGAP100, DGAP125, DGAP133, DGAP134, DGAP154, DGAP159, DGAP167, DGAP169, DGAP170, DGAP220, ROC17 |
| Exiguobacterium oxidotolerans | High catalase activity may disable immune defenses depending on peroxide. | DGAP127, DGAP225 |
Stealth viruses are frequent matches to chromosome anomalies with about 35 matching sequences. Stealth viruses are mostly derivatives of herpes viruses such as cytomegalovirus (CMV) that emerge in immunosuppressed patients or have other mechanisms to avoid recognition by the immune response. CMV is not a well-known teratogen, even though CMV is a major threat to infants. First trimester CMV infection can cause severe cerebral abnormalities followed by neurologic symptoms [14]. CMV is also a common cause of congenital deafness and can cause visual abnormalities. 27 of the 48 neurodevelopmental patients had hearing loss or deafness. Stealth virus 1 refers to Simian cytomegalovirus (strain Colburn) African green monkey cytomegalovirus, but closely related viruses with up to 95% sequence identity have been isolated from human patients. Herpes simplex virus is another stealth virus that directly infects the central nervous system. Herpes simplex can cause seizures (reported for 9 patients).
HTLV-1 is a retrovirus that causes an immune mediated disease of the nervous system affecting the spinal cord and peripheral nerves. Neural fetal cells are more susceptible to HTLV-1 infection than mature neurons.
Patient DGAP159 has the strongest homology to N. meningitidis (Figure 2). N. meningitidis causes bacterial meningitis and is a known cause of neurodevelopmental defects. Signs and symptoms in patient DGAP159 are consistent with residual effects of bacterial meningitis such as hearing loss, developmental delay, speech failure and visual problems.
DNA from microbes not classified as teratogens can also affect fetal development
Not only known teratogens, but almost all microbial DNA found in abnormal human chromosomes in congenital neurologic disorders can have profound effects on a fetus or neonate (Table 2). Figure 4 implies that any source of viral or microbial foreign DNA may contribute to developmental errors. Multiple matches may represent homologies to human DNA among stretches of viral and bacterial DNA. Homologies found appear independent of whether microbes can insert into human DNA. HIV, HPV16, HTLV-1, k. pneumoniae, ralstonia solanacearum, s. aureus, and stealth viruses are all found in at least 10 patients. 38 of 48 rearrangements include sequences that match one or more viruses, and 44 match bacterial sequences. In most chromosomal anomalies, multiple infections match a given anomalous DNA sequence. It is likely that most or all of these infections can interfere with neurodevelopment (Tables 1-2). Figure 4 includes endogenous retroviral sequences. Most of these endogenous sequences represent crippled infections that cannot travel between cells because they lack env genes needed to exit infected cells. However, HERV sequences are numerous and retrotransposons can still interfere with recombination and DNA repair within their home cell.
Deleted segments in familial chromosome anomalies point toward a general mechanism for infection as a cause of neurodevelopmental disorders
Effects of balanced translocations segregate only somewhat with the presence of neurodevelopmental disorders. These discrepancies emphasize the need to consider the genome in 3 dimensions [8]. Moreover, there may also be other more cryptic chromosome abnormalities and that whole genome sequencing of the family may be necessary [3]. Members of some families carry balanced chromosomal translocations but do not have signs and symptoms of neurodevelopmental disease. In three of four families with familial balanced chromosomal translocations, patient specific unbalanced deletions were also found [3] but the results did not overlap any database of human reference genomes. Unbalanced deletions were subjected to analysis for infection homology but inversions, inverted duplications, insertions and additional balanced translocations were also present. Unbalanced deletions in the three families were strongly homologous to infections or other foreign DNAs (Figure 6). In agreement with results in Table 1, critical genes linked to epigenetic modifications were among those interrupted by these short cryptic rearrangements.
Figure 6
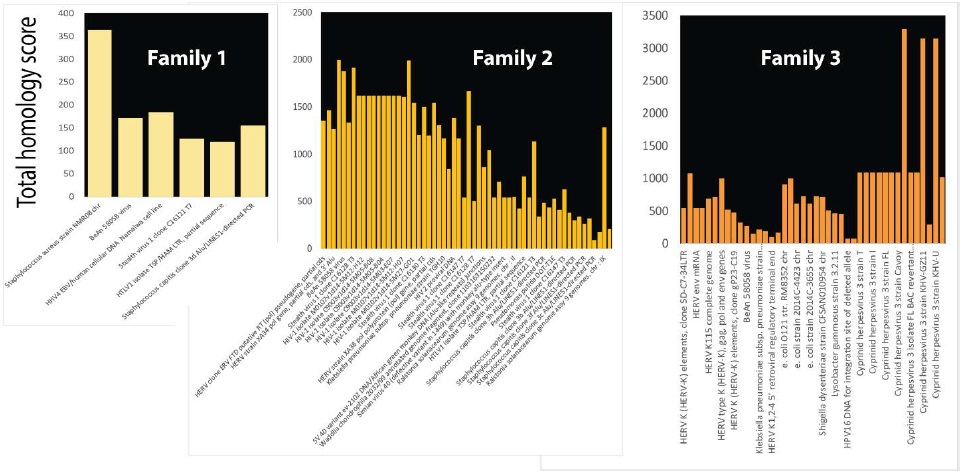
Figure 6: Unbalanced deletions present in an affected family are homologous to microorganisms. Family members affected by intellectual disability in families with apparently balanced translocations have other chromosome abnormalities including unbalanced deletions, insertions, etc [3]. Deletions are shown as homologous to bacterial and viral DNA, but insertions were also found to have similar homologies. Total homology scores for bacterial and viral homologies in each of the three affected patients are plotted.
Microbial homologies in a recurrent translocation
De novo translocations t(11;22) have so far only been detected during spermatogenesis and not during other processes. These translocations have been attributed to palindromic structures that induce genomic instability [2]. To determine whether these structure might arise because microbial infection interferes with normal chromatin structures, homology testing of sequences in the recurrent breakpoint t(8;22)(q24.13;q11.21) [2] was conducted. Figure 7 shows strong homology to bacterial and viral sequences in the breakpoint region. Sequences from an unaffected mother carry a balanced translocation rearrangement [2] with homologies to more diverse microbes than are present in affected cases (Figure 7). This supports the idea that parts of a chromosome segment with homologies to infection have been deleted in affected family members.
Figure 7
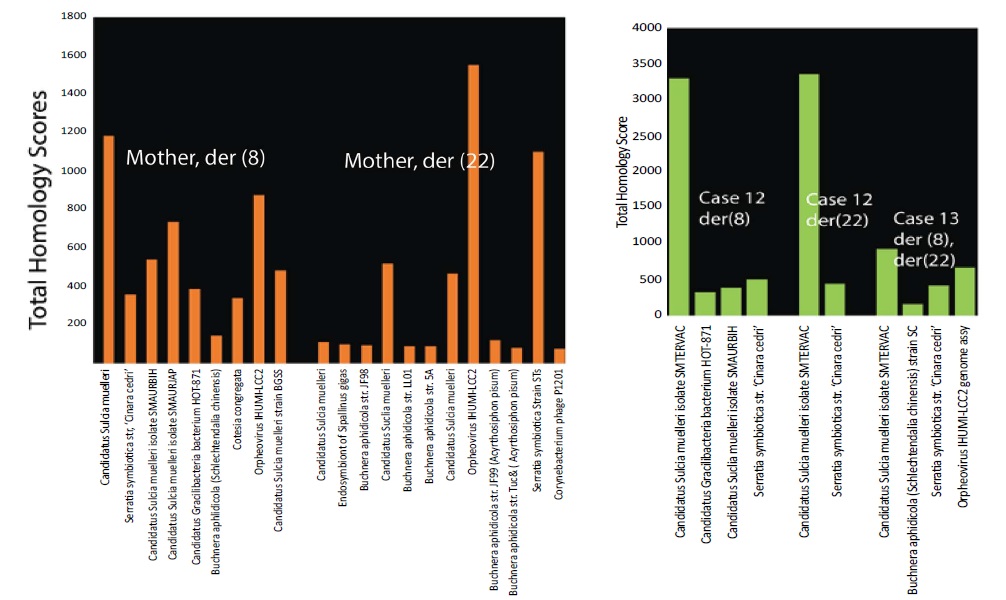
Figure 7: Microbial homologies in cases from a maternal recurrent translocation. Parental translocation t (8;22) in Family FHU13-027 from Mishra and colleagues [2] is homologous to microorganisms as shown for der (8) and for der (22) separated by space in the figure. The sequence is from a normal healthy mother who inherited from a female proband the balanced translocation t (8;22). More limited sets of micro-organisms are present in affected case 12 and case 13 (green bars) implying that DNA homologous to the other microorganisms has been lost.
A model for infection interference in neurodevelopment
Infection DNA occurs in neurodevelopmental disorders that undergo autosomal dominant inheritance so microbial DNA must be present in gametes from one parent. The mechanism proposed in Figure 8 is based on data showing significant homologies with microbial infections on multiple human chromosomes. Large amounts of foreign, infection DNA present during human meiosis with its many double strand breaks and the most active period of recombination produce defective gametes.
Figure 8
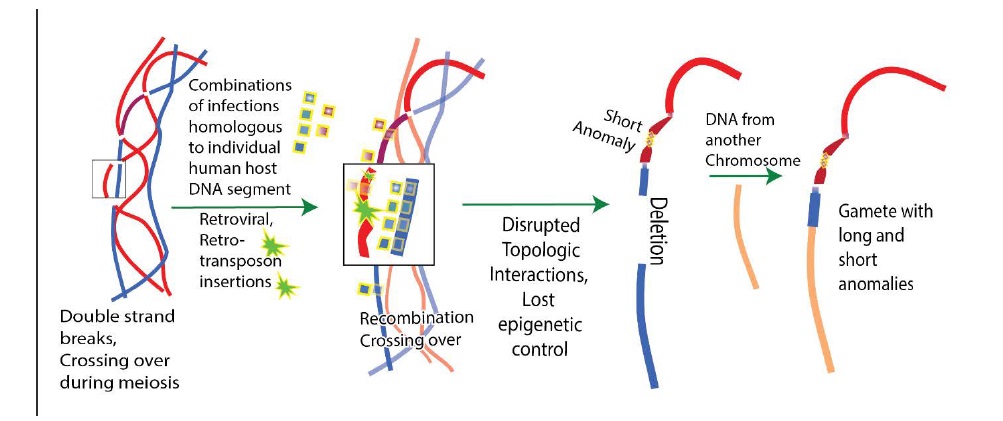
Figure 8: Model for interference with meiosis by DNA from infections leading to neurodevelopmental disorders. After duplication of parental chromosomes prior to generation of gametes, DNA from infections associates with strands of DNA in the many places including repetitive sequences of human DNA closely matching infection DNA. In addition, retroviruses and retrotransposons may integrate their DNA. These events interfere with reductive cell division, topological relationships among chromosomes, epigenetic regulation and high-fidelity break repair. Interference from microorganisms may favor the illegitimate combinations due to palindromes reported by Mishra and co-workers. Hundreds of DNA breaks occur during meiosis and incorrect repair of these breaks is known to occur [1], causing chromosome anomalies such as deletions (shown). Clustered genes responsible for linked functions and epigenetic regulation in neurodevelopment are lost or displaced. Other chromosome segments with microbial homology do not contain identified genes but may be essential control regions or they may be involved in chromatin structures. There are about one million Alu repeats alone in the human genome and many match sets of infections in individual neurodevelopment disorders. More private matches also occur. When combined with effects from other microorganisms this can cause severe disruption of base pairing in gamete generation.
The resemblance of foreign microbial DNA to the host background DNA may be a major factor in selection of infection and in human ability to clear the infection. Only one rare defective gamete is shown in Figure 8 but the male generates four gametes during meiosis beginning at puberty. Only one gamete survives in the female because three polar bodies are generated. Both balanced and unbalanced translocations can give rise to chromosome anomalies such as deletions and insertions because infection DNA can insert itself (e.g. exogenous or endogenous retroviruses) or interfere with break repairs during meiosis. The presence of a preexisting balanced chromosomal translocation in the family disrupts normal chromatin structure, producing both affected and unaffected members [3]. The familial translocation increases the chances of generating a defective gamete during meiosis.
Tests for artifacts in matches to human-microbial DNA
Genome rearrangements for patient data in Figure 4 [1]
produced 1986 matches with E
The 1986 microbial homologues to human DNA near breakpoints involved in neurodevelopmental disorders were composed of 126 different viral or bacterial sequences. 24 different strains of Neisseria meningitidis in patient DGAP159 had 100% identity to human genomic rearrangements with total scores of 385 to 637 and expected values of 8e-13. The 25th N. meningitidis strain was 98% identical. Human matches included several 5’UTR like sequences, elongation factor alpha, isopentenyl pyrophosphate transferase-like protein, NADH dehydrogenases. Clostridium specifically matched sections of human ATPases, and ATP synthases, mitochondrial G elongation factor, and heat shock protein hsp70 (mRNA). A 1004 bp fragment of waddlia chondrophila occurred 32 times near neurodevelopmental breakpoints and had full-length 89% homology to human calcyclin binding protein (E=0.0). Waddlia chondrophila also had strong matches to human caspase recruitment domain and human cloned fragments. Another 55 of 126 viral / bacterial sequences were homologous to Alu sequences. Alu sequences are largely inactive retrotransposons, but it is possible that some of the homologies detected between humans and microbes are actually due to insertions from Alu or other repetitive sequences. Neuronal progenitors may support de novo retrotransposition in response to the environment or maternal factors [16]. Both non-Alu and Alu regions account for 90% (113 of 126) of the sequences homologous to microbes and present near breakpoints involved in human neurodevelopmental disorders.
Tests for DNA sequence artifacts
To further test the possibility that some versions of these microbial sequences were sequencing artifacts or were contaminated by human genomes, microbial genomes represented in the Figures and other arbitrarily selected microbial genomes were tested for homology to human genomes. Results of these reversed-analyses make it unlikely that the human-microbial matches are errors or artifacts. Substantial similarities between partial and complete microbial DNAs and human DNAs were universal and helped develop the model in Figure 7.
Homologies between multiple strains of the same microbe and all human sequences were tested. Homologies to human sequences were found across multiple strains of the same microorganism (Table 3). In other cases, homology to some human gene product was found but there was not sufficient data available to compare multiple versions of the same microorganism. In these cases, the human gene was tested against all bacteria, and viruses. There were hundreds of significant matches. The complete clostridium botulinum genome had significant homology to human enzyme CDKL2 (86%) on chromosome 4. The region on chromosome 4: 579,587-75,632,298 spanned 52712 bp and was filled with over 50 Alu repeats, including multiple versions of AluS, AluY, and AluJ. Clostridium itself however did not have homology to Alu repeats. Clostridium also had significant homology with long stretches on other chromosomes including chromosomes 2, 3, 10, 15, 1, 5 with 74% homology over about 2853 bp E=0.0. Similarly, the full genome of waddlia chondrophila (2.1 million bp) was homologous to multiple human genes (Table 3).
Table 3: Independent evidence that microbial genomes have regions of homology to human DNA as predicted by results.
| Microbe | Human chromosome homologies | Length of homology (bp) | %homology, E value |
|---|---|---|---|
| N. gonorrhoeae strain 1090 | NotI sites | 233-345 | 100%, 5e-89 |
| N. gonorrhoeae WHO-L genome (GenBank: LT591901.1) | 700 bp on Chr 3, 15, X and 3 | 700 | 99-100% |
| N. gonorrhoeae strain 1090 | Clone image | 519 | 79% |
| S aureus | Cu transport ATPase | 1198 | 98%, E=0.0 |
| NC_007795.1 Staphylococcus aureus subsp. aureus | P143 mRNA | 642 | 70%, E=9e-58 |
| S capitis | ABC8 | 218 | 71%, 5e-15 |
| MRSA | Succinate CoA ligase, Chr13 | 769 | 67% |
| Ralstonia solanacearum | s-adenosyl-homocysteine hydrolase >NM_001242673.1 Homo sapiens adenosylhomocysteinase like 1 (AHCYL1), transcript variant 2, mRNA (Transcript variant 2 easily produced >100 significant matches to other microbial sequences) | 791 | 69%. 2e-73 |
| Pseudomonas putida strain IEC33019 chromosome, complete genome NCBI Reference Sequence: NZ_CP016634.1 vs strain T1E (CP003734.1) | Many hundreds of homologies e.g. homo sapiens sequence around Not1 site clone HSJ-DM24RS | 674 bp | 92%, E=0.0 |
| C botulinum | HSP70 member 9 | 1066 | 65% E=5e-54 |
| C botulinum BrDura | Human cDNA | 692 | 83%, E=0.0 |
| Waddlia chondrophila (CP001928.1) whole genome 2.1 million bp | Phosphoglycerate dehydrogenase, fumarate hydratase elongation factor alpha hsp70 family member 9 aldehyde dehydrogenase |
734 1034 992 1548 1168 |
66% E=9e-71 67% E=3e-63 71%E=1e-112 65% E=1e-68 64% E=1e-30 |
| 67% BeAn 58058 virus NM_001165931.1 9 Closely cotia virus and other pox viruses) ribonucleotide reductase > 100 significant matches to cotia virus (90% homology), Volepox, monkeypox, cowpox, and vaccinia virus e.g. 675/939 bp(72%) E=4e-136 | Ribonucleotide regulatory subunit (Homo sapiens ribonucleotide reductase regulatory subunit M2 (RRM2), transcript variant 1, mRNA) | 870 | 68%, 8e-69 |
| Cowpox, KY369926.1 Description Cowpox virus strain Kostroma_2015, complete genome Molecule type nucleic acid Query Length 223595 |
At least 100 homologies. Homologies to ribonucleotide reductase, z-protein mRNA, transmembrane BAX inhibitor motif, EF hand domain containing EFHC2 | 68-78% homology | 2279 bp at 69% homology (E=0.0) |
| HIV-1 28 different isolates | Alu homology bp7300 to 8000 | Up to 98% identity for all 28 sequences | |
| HTLV1 J02029 vs HTLV1/HAM Long terminal repeat | Homologous to hydroxysteroid dehydrogenase like 1 variant, mRNA for hGLI2 | 97% homology | 2162 bp at 97% homology (E=0.0) |
Retroviral infections identified in Fig 1 are known to insert their sequences in human DNA or have other methods of infection and transmission, such as by inhibiting key protective mechanisms. For example, HIV-1 destroys helper T-cells after uptake by a specific receptor. Like other retroviruses, HIV-1 DNA becomes incorporated into host cells. HIV-1 and other retroviral insertions were found in the congenital abnormalities in neurodevelopmental disorders. HIV-1 isolate SC007 from India, partial genome (GenBank: KY713228.1) had 85% identity over 686 bases to an IgG2 lambda antibody and 82% homology to a 412 bp stretch of human DNA surrounding a rare NotI restriction site. HIV homologies were very commonly found among human sequences of neurodevelopmental disorders. To test for human sequence contaminants in these matching HIV sequences, an Alu homologous region of the HIV-1 genome (bp 7300 to 9000) in 28 different HIV-1 isolates in an HIV sequence compendium were compared. All 28 HIV sequences had matches to the same region of human DNA, with up to 98% identity to the human sequences. The best match was to a LINCRNA on chr11:74,929,289-74,929,412. In contrast, only one sequence of zika virus matched humans in a short segment of human-like DNA on the 3’ end. This homology was not found in 20 additional zika virus sequences so zika virus was not considered further.
It is almost impossible to completely exclude the possibility of sequencing artifacts or contamination of microbial sequences with human sequences. However rather than reflecting widespread, wholesale error due to human DNA contamination in many laboratories over many years, the vast majority of microbial homologies more likely suggest that infection sequences have been selected because they are homologous to regions of human DNA. This may be a driving force behind the much slower evolution of Alu DNA.
Discussion
Long stretches of DNA in many infections match repetitive human DNA sequences which occur hundreds of thousands of times. Individual microorganisms also match nonrepetitive sequences. Human infections may be selected for and initially tolerated because of these matches. Infections can cause mutations by interfering with the most active period of recombination which occurs during the generation of gametes, an ongoing process in males. Infections such as exogenous or endogenous retroviruses are known to insert into DNA hotspots [8]. Human DNA sequences that closely match infection DNA generate chromosomal anomalies because similarities between microbial and human DNA cause interference with meiosis (Fig. 7). Thus, the genetic background of an individual may be a key factor in determining the susceptibility to infection and to the effects of infection. At the genetic level, this suggests selective pressures for infections to develop and use genes that are similar to human genes and to silence or mutate microbial genes that are immunogenic. Infection genomes evolve rapidly on transfer to a new host [17]. The presence of genes in infections that have long stretches of identity with human genes makes the infection more difficult to recognize as non-self. For example, there is no state of immunity to N. gonorrhoeae. One reason for this is that there are long stretches of N. gonorrhoeae DNA that are almost identical to human DNA. Alu sequences and other repetitive elements are thought to underlie some diseases by interfering with correct homologous recombination as in hereditary colon cancer [18] or abnormal splicing. Why this does not always occur is not well understood. The presence of infection DNA that is homologous to multiple, long stretches of human DNA may mask proper recombination sites and encourage this abnormal behavior.
Neurons and cells in the immune system are interacting systems, sensing and adapting to their common environment. These interactions prevent multiple pathological changes in many disorders [19]. Many genes that are implicated in neurodevelopmental diseases, reflect the strong relationship between the immune system and the nervous system. It was always possible to find functions within the immune system for genes involved in neurodevelopmental disorders (Figs 1 and 4). Damage to genes essential to prevent infection leads to more global developmental neurologic defects including intellectual disability. These microbial homologies include known teratogens. Analysis of mutations within clusters of genes deleted in neurodevelopmental disorders predict loss of brain-circulatory barriers, facilitating infections. Damage to cellular genes essential for the immune related function of autophagy may lead to abnormal pruning of neural connections during postnatal development.
Aggregated gene damage accounts for immune, circulatory, and structural deficits that accompany neurologic deficits. Other gene losses listed in Table 1 and in deleted chromosome segments (Figure 1) account for deficits in cardiac function, hearing, bone structure, pain perception, skull size, muscle tone and many other nonneurologic signs of neurodevelopmental disorders.
The arrangement of genes in clusters converging on the same biological process may simplify the regulation and coordination between neurons and other genes during neurodevelopment and neuroplasticity. Genes that are required for related functions, requiring coordinated regulation have been shown to be organized into individual topologically associated domains [20]. Neurons are intimately connected to chromatin architecture and the epigenetic landscape [21]. A disadvantage of the clustered arrangement of co-regulated accessory genes is that homology to microbial infections or other causes of chromosome anomalies anywhere in the cluster can then ruin complex critical neurological processes. Epigenetic regulators that affect multiple functions required by the same process make their mutation especially critical. Longer range developmental interactions in chromosome regions exacerbate the effects of infection. Mutations or deletions (Figure 1 and Table 1) show that this effect can occur in neurodevelopmental disorders. Functions that must be synchronized during neurodevelopment are grouped together on the same chromosome region and can be lost together. This is consistent with the idea that the organization of crossover chiasmata during meiosis allows the coordinated evolution and control of gene sets required by the same essential process.
Microbial DNA sequences are unlikely to be contaminants or sequencing artifacts. They are all found connected to human DNA in disease chromosomes; multiple sequences from different laboratories are all homologous to the same Alu sequence. Alu element-containing RNA polymerase II transcripts (AluRNAs) determine nucleolar structure and rRNA synthesis and may regulate nucleolar assembly as the cell cycle progresses and as the cell adapts to external signals [22]. HIV-1 integration occurs with some preference near or within Alu repeats, regardless of whether they occur in introns or exons [23].
The patients indicated in Fig. 3 have different disorders with different chromosome anomalies. This variability and the rarity of the anomalies makes these disorders difficult to study by conventional approaches. The techniques used here might eventually help predict the lifelong susceptibility to a given infection of an individual. However, the methods used suffer from the inability to absolutely identify one infection and to distinguish infection by one microorganism from multiple infections.
Conclusion
- DNAs in some congenital neurodevelopmental disorders closely matches DNA in multiple infections that extend over long linear stretches of human DNA and often involve repetitive human DNA sequences. The affected sequences are shown to exist as linear clusters of genes closely spaced in two dimensions.
- Interference from infection can delete or damage human gene clusters and epigenetic regulators that coordinate neurodevelopment. This genomic interference accounts for immune, circulatory, and structural deficits that accompany neurologic deficits.
- Neurodevelopmental disorders are proposed to begin when parental infections cause insertions or interfere with meiosis at both repetitive and unique human DNA sequences.
- Congenital neurodevelopmental disorders are thus viewed as resulting from an assault on human DNA by microorganisms and an example of the selection of infecting microorganisms based on their similarity to host DNA.
Funding
This research received no external funding but an Incentive award to BF was used for support.
Acknowledgment
This work is based on the outstanding DNA sequence information from investigators listed in the references and the author is grateful to them for making this information available. It is a pleasure to acknowledge the inspiration, challenge and stimulation provided by the Department of Biochemistry and Molecular Genetics and the College of Medicine at UIC.
Conflicts of Interest
The author declares no conflict of interest.

View pdf
Download pdf
There is no references
