Journal Name: Journal of Biomedical Research and Reviews
Article Type: Research
Received date: 16-October-2020
Accepted date: 11-November-2020
Published date: 18-November-2020
Citation: Wu W, Fang C, Zhang C, Hu N, Wang L (2020) Identification of 10 Important Genes with Poor Prognosis in Non-Small Cell Lung Cancer through Bioinformatical Analysis. J Biomed Res Rev Vol: 3, Issu: 2 (41-50).
Copyright: © 2020 Wang L. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Objective: The lung cancer has become the most lethal cause of cancer-related death in China and is responsible for more than 1 million deaths all over the world every year, especially non-small cell lung cancer (NSCLC). Although great advance in pharmaceutical therapies for lung cancer patients, the overall survival is still poor. It is necessary to find out the effective biomarkers in order to improve and predict the prognosis of lung cancer patients. The integrated bioinformatical analysis, as a useful tool to dig up the valuable clues, can be applied to search new effective therapeutic targets.
Methods: In this work, we utilized four NSCLC datasets (GSE18842, GSE31210, GSE33532 and GSE101929) from Gene Expression Omnibus (GEO) to analyze. We totally found that there were 162 differentially expressed genes (DEGs) in these four datasets, including 41 up-regulated genes and 121 down-regulated genes in NSCLC tissues. The analysis of gene ontology (GO) enrichment and Kyoto encyclopedia of genes and genomes (KEGG) pathway was done by Database for Annotation, Visualization and Integrated Discovery (DAVID) software. Then, we identified 10 core oncogenes by constructing protein-protein interaction (PPI) network. Last, we further analyzed the 10 core oncogenes through Kaplan Meier plotter online database and Gene Expression Profiling Interactive Analysis (GEPIA) respectively.
Results: We discovered 10 key oncogenes which were associated with the progression and poor prognosis for NSCLC, including ANLN, CCNA2, CDCA7, DEPDC1, DLGAP5, HMMR, KIAA0101, RRM2, TOP2A, and UBE2T.
Conclusion: These 10 genes can be served as the therapeutic targets and useful prognostic biomarkers for NSCLC treatment.
Keywords
Non-small cell lung cancer; Bioinformatical analysis; Prognostic biomarkers; Differentially expressed genes; Therapeutic targets.
Abstract
Objective: The lung cancer has become the most lethal cause of cancer-related death in China and is responsible for more than 1 million deaths all over the world every year, especially non-small cell lung cancer (NSCLC). Although great advance in pharmaceutical therapies for lung cancer patients, the overall survival is still poor. It is necessary to find out the effective biomarkers in order to improve and predict the prognosis of lung cancer patients. The integrated bioinformatical analysis, as a useful tool to dig up the valuable clues, can be applied to search new effective therapeutic targets.
Methods: In this work, we utilized four NSCLC datasets (GSE18842, GSE31210, GSE33532 and GSE101929) from Gene Expression Omnibus (GEO) to analyze. We totally found that there were 162 differentially expressed genes (DEGs) in these four datasets, including 41 up-regulated genes and 121 down-regulated genes in NSCLC tissues. The analysis of gene ontology (GO) enrichment and Kyoto encyclopedia of genes and genomes (KEGG) pathway was done by Database for Annotation, Visualization and Integrated Discovery (DAVID) software. Then, we identified 10 core oncogenes by constructing protein-protein interaction (PPI) network. Last, we further analyzed the 10 core oncogenes through Kaplan Meier plotter online database and Gene Expression Profiling Interactive Analysis (GEPIA) respectively.
Results: We discovered 10 key oncogenes which were associated with the progression and poor prognosis for NSCLC, including ANLN, CCNA2, CDCA7, DEPDC1, DLGAP5, HMMR, KIAA0101, RRM2, TOP2A, and UBE2T.
Conclusion: These 10 genes can be served as the therapeutic targets and useful prognostic biomarkers for NSCLC treatment.
Keywords
Non-small cell lung cancer; Bioinformatical analysis; Prognostic biomarkers; Differentially expressed genes; Therapeutic targets.
Introduction
The lung cancer, which is the leading cause of cancer-related death in China and is responsible for more than 1 million deaths all over the world every year [1], can be divided two classes: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC accounts for approximately 85% of all lung cancer cases, including adenocarcinoma, squamous cell carcinoma and large cell carcinoma. Nowadays, although great advance in pharmaceutical therapies for lung cancer patients, the overall survival is still poor. NSCLC has become the most lethal human cancer. Hence, it is necessary to find out the effective therapeutic targets in order to improve the prognosis of lung cancer patients.
Gene chip, as a proven technique, could make many slice data be produced and stored in public databases [2]. Therefore, we can explore a large number of valuable clues via these data. Meanwhile, the integrated bioinformatic results also can help us to further study and discover the potential mechanism. In order to further discover the novel targets for treating NSCLC, we applied the public gene chip databases to carry on data mining. Through this study, we could find out some new candidate therapeutic targets for NSCLC, also it is useful for improving the survival and the quality of life.
In the present study, as shown in figure 1, we chose 4 databases related with non-small cell lung cancer from Gene Expression Omnibus (GEO), including GSE18842, GSE31210, GSE33532 and GSE101929. First, we found that there were 162 differentially expressed genes (DEGs) in these four databases above, including 41 up-regulated genes and 121 down-regulated genes in NSCLC tissues. Then, we did some other bioinformatic analyses and identified 10 core genes by establishing protein-protein interaction (PPI) network. In order to confirm the important role of these 10 core genes in NSCLC, we further analyzed the survival curve and the DEGs expression between NSCLC tissues and normal lung tissues through Kaplan Meier plotter online database and Gene Expression Profiling Interactive Analysis (GEPIA) respectively. Taken above, these 10 DEGs were all related with the prognosis of NSCLC. In conclusion, our bioinformatic study provides some additional useful biomarkers for NSCLC patients. These biomarkers can be considered as candidate therapeutic targets for NSCLC, and the results also supply some ideas for our further study.
Materials and Methods
Data source and preprocessing
NCBI-GEO (https://www.ncbi.nlm.nih.gov/geo/) was selected for our research, which is a free public database of microarray/gene profile. We used the key words (‘non-small cell lung cancer’ [All Fields] OR ‘lung adenocarcinomas’ [All Fields]) AND (‘human’ [Organism]) AND (‘Expression profiling by array’ [Filter]) to select related datasets. Next, we screened four gene expression profiles (including GSE18842, GSE31210, GSE33532 and GSE101929) according to the following inclusion criteria: a. Human NSCLC tissues, not cell lines; b. Normal lung tissues used as controls; c. The total sample numbers, containing tumor tissues and normal tissues, are over 50; d. These datasets have the same Platform in order to process the data easily. These four gene profiles we selected were all on account of GPL570 Platform. GSE18842 contained 46 NSCLC tissues and 45 normal lung tissues, GSE31210 included 226 NSCLC tissues and 20 normal lung tissues, GSE33532 covered 80 NSCLC tissues and 20 normal lung tissues, and GSE101929 incorporated 32 NSCLC tissues and 34 normal lung tissues.
Screening of differentially expressed genes (DEGs)
The DEGs between NSCLC tissues and normal lung tissues were screened by using the GEO2R online tools. The fold change value (FC) obtained for each genes was indicated as logFC in order to normalize the data derived from the same microarray platform [3]. We considered DEGs as |logFC| >2 and adjust P value < 0.05. Venn software online (http://bioinformatics.psb.ugent.be/webtools/Venn/) was used to analyze the DEGs among the above four datasets via checking the raw data in TXT format. In the present study, the DEGs with log FC > 2 was considered as an up-regulated gene, and the DEGs with log FC < -2 was regarded as a downregulated gene.
DEGs gene ontology (GO) enrichment and Kyoto encyclopedia of genes and genomes (KEGG) pathway analyses
After screening the DEGs from the above four datasets, we performed the GO enrichment and KEGG pathway analyses using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf. gov/tools.jsp), which is designed to identify a huge number of genes or proteins function [4]. GO analysis is used to integrate annotation data and provide tools access to all the data provided by the study, and identify unique biological properties of these datasets [5]. KEGG can integrate the currently known protein interaction network information, including metabolism, genetic information processing, environmental information related processes, and cell physiological process, etc [6]. We used DAVID to perform biological analyses of DEGs and visualize the DEGs enrichment of biological processes (BP), molecular functions (MF), cellular components (CC) and pathways. P<0.05 was considered as significant difference.
Protein-protein interaction (PPI) network analysis
PPI network analysis was performed for the identified DEGs by using Search Tool for the Retrieval of Interacting Genes (STRING) (https://string-db.org/), which is an online software of interactions of genes and proteins. The PPI network could be visualized by Cytoscape in order to examine the potential correlation between the DEGs (maximum number of interactors=0 and confidence score ≥0.4) [7]. Besides, the Molecular Complex Detection (MCODE) app in Cytoscape was used to analyze the modules of the PPI network (degree cutoff=2, max. Depth=100, κ-core=2, and node score cutoff=0.2) [8].
Analyzing overall survival and RNA sequencing expression of core genes
Kaplan Meier-plotter (https://kmplot.com/analysis/) is a widely used website tool for illustrating the relationship between patients’ overall survival and gene expression levels of DEGs based on EGA, TCGA and GEO [9]. In this study, we acquired core genes corrected with the progression of NSCLC through the PPI network analysis. The correlation between core genes expression and survival in lung cancer was analyzed by Kaplan Meier-plotter. The hazard ratio (HR) with 95% confidence intervals and log-rank P value were also computed and showed on the plot. In order to validate the important of these core genes, we next used the GEPIA website (http://gepia.cancer-pku.cn/) to analyze the RNA sequencing expression data according to thousands of samples from the GTEx projects and TCGA [10], including lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC).
Figure 1
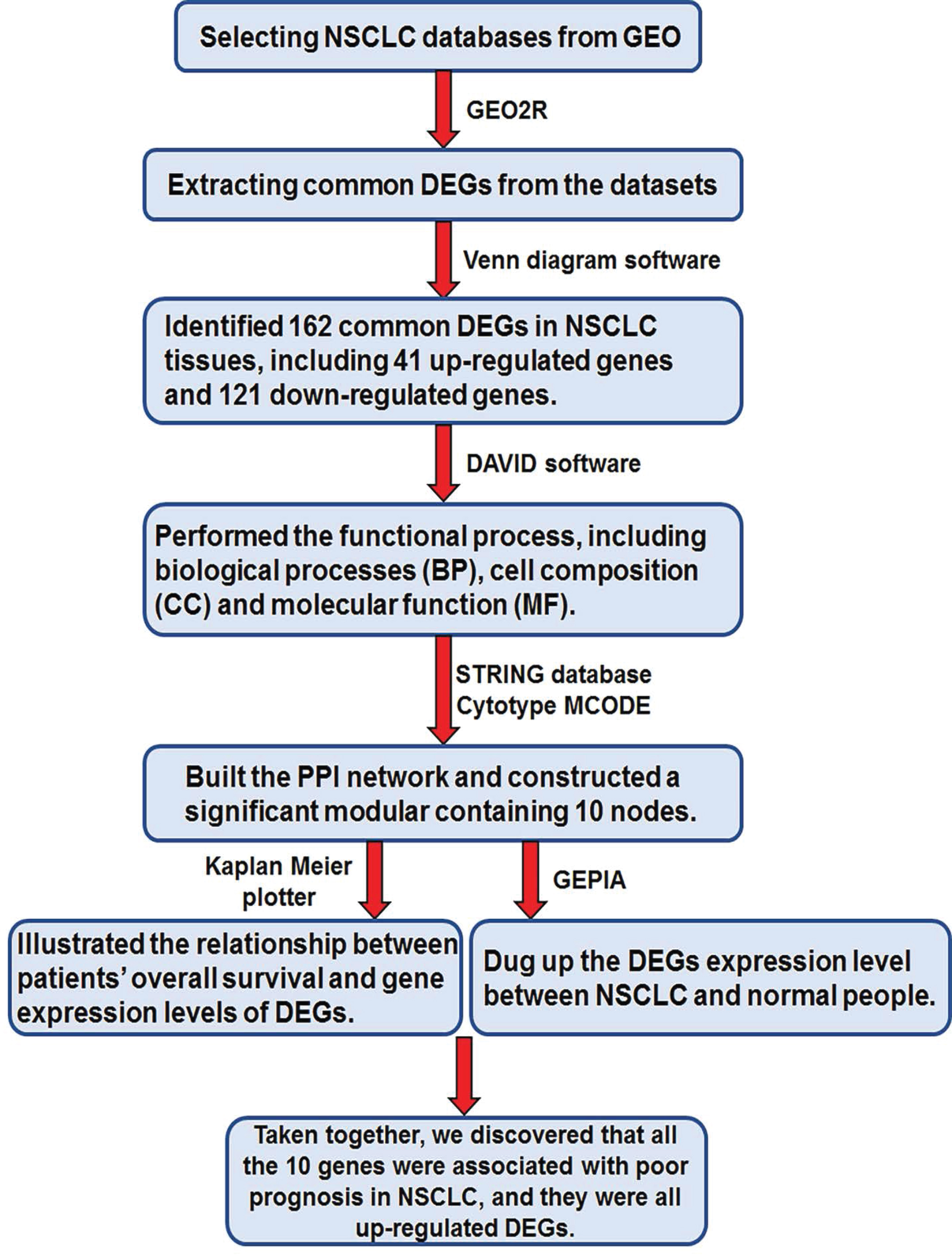
Figure 1: The process of the work.
Results
Screening of DEGs between NSCLC tissues and normal lung tissues
There were together 384 NSCLC tissues and 119 normal lung tissues in these four datasets that we chose to study. The up-regulated DEGs were statistically significant as logFC > 2 and P value < 0.05, while the down-regulated DEGs were statistically significant as logFC < -2 and P value < 0.05. Through GEO2R online tools, a total of 772, 443, 610, and 926 DEGs were extracted from GSE18842, GSE31210, GSE33532 and GSE101929, respectively. Among these DEGs, 317 up-regulated DEGs and 455 down-regulated DEGs were included in GSE18842; 171 up-regulated DEGs and 272 down-regulated DEGs were contained in GSE31210; 201 up-regulated DEGs and 409 down-regulated DEGs were covered in GSE33532; 318 up-regulated DEGs and 608 down-regulated DEGs were incorporated in GSE101929. Then, we screened the common DEGs in these four datasets by applying Venn diagram software online. Finally, we identified 162 common DEGs in NSCLC tissues, including 41 up-regulated genes and 121 down-regulated genes (Figure 2 and Table 1).
Figure 2
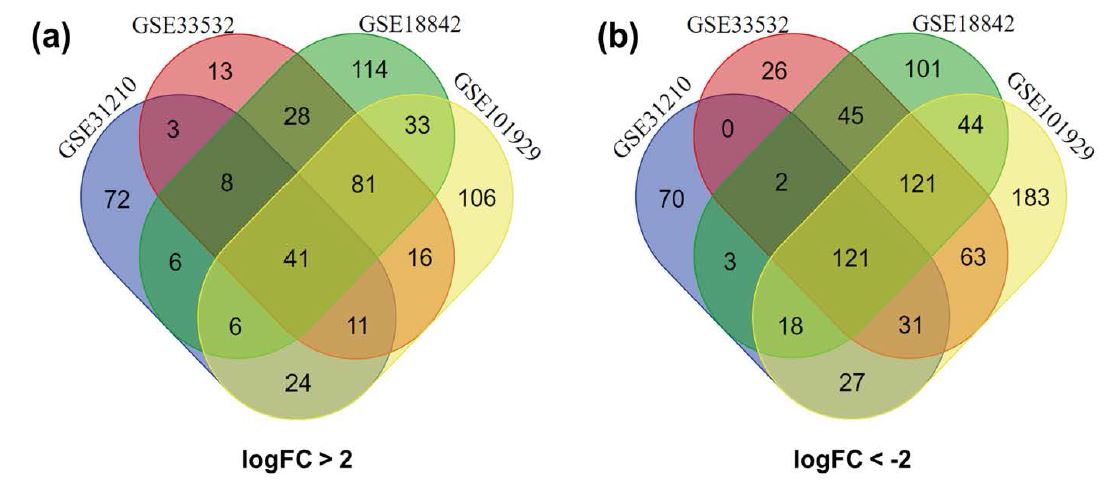
Figure 2: Screening of 162 common DEGs from four gene expression datasets (GSE18842, GSE31210, GSE33532 and GSE101929) by applying Venn software online. Different color meant different datasets. (a), 41 common up-regulated DEGs in the four datasets (logFC>2); (b), 121 common down-regulated DEGs in the four datasets (logFC<-2).
Analyzing of the DEGs GO enrichment
In our study, all 162 DEGs were analyzed by DAVID software in order to perform the functional process. The results were shown in figure 3 and table 2. In this part, we only summarized the top 5 different functional process: a. in the biological processes (BP) section, the up-regulated DEGs were mainly involved in collagen catabolic process, extracellular matrix disassembly, collagen fibril organization, sensory perception of sound, and proteolysis, while the down-regulated DEGs in angiogenesis, vasculogenesis, cell surface receptor signaling pathway, receptor internalization and vasoconstriction; b. in the cell composition (CC) part, the up-regulated DEGs were enriched in proteinaceous extracellular matrix, collagen trimer, extracellular region, and cytoplasm, while the down-regulated DEGs in integral component of plasma membrane, integral component of membrane, membrane raft, plasma membrane, and external side of plasma membrane; c. in the molecular function (MF) aspect, the down-regulated DEGs were particularly focused on receptor activity, heparin binding, ion channel binding, Ras guanyl-nucleotide exchange factor activity and angiotensin type II receptor activity, while up-regulated DEGs in no significant difference.
Analyzing of the DEGs KEGG pathways
In this study, the DEGs KEGG pathways were also performed by DAVID software. As shown in table 3, the results indicated that the DEGs were mainly enriched in ECM-receptor interaction, cell adhesion molecules, leukocyte transendothelial migration, protein digestion and absorption, PPAR signaling pathway, adrenergic signaling in cardiomyocytes and neuroactive ligand-receptor interaction.
Table 1: All 162 common DEGs were screened from four gene expression datasets, including 41 up-regulated genes and 121 down-regulated genes.
| DEGs | Name of genes |
|---|---|
| Up-regulated DEGs | CDH3 IGF2BP3 HMGB3 CRABP2 CXCL13 AKR1B10 COL1A1 ADAMDEC1 ANLN RRM2 TOP2A GJB2 TFAP2A DLGAP5 ARNTL2 FERMT1 HMMR ANKRD22 TMPRSS4 HS6ST2 SPP1 NMU SIX1 COL10A1 LRRC15 GPR87 CDCA7 COL11A1 PLPP2 CTHRC1 KIAA0101 GREM1 CCNA2 CP MMP1 MMP12 UBE2T MMP9 DEPDC1 MMP11 FAM83A |
| Down-regulated DEGs | HBA2///HBA1 RTKN2 EMCN SOX7 GPIHBP1 KCNT2 MFAP4 PEBP4 SLC6A4 PECAM1 KCNK3 MMRN2 NOSTRIN NCKAP5 OGN SCARA5 CLDN5 BTNL9 IGSF10 SCGB1A1 CDO1 HIGD1B CA4 SDPR TEK GRK5 ID4 EXOSC7///CLEC3B DACH1 LOC100653057///CES1 FAM150B ACKR1 STXBP6 LYVE1 ADAMTS8 GDF10 LEPROT///LEPR AKAP12 CD36 FAM162B GPD1 HSPA12B ROBO4 SPTBN1 CALCRL CAV1 RASIP1 PPBP JAM2 PTPRB FOXF1 ACADL ANKRD29 PIR-FIGF///FIGF AQP4 NEBL MT1M TNNC1 MCEMP1 HBB SERTM1 SELE FHL1 CPB2 SSTR1 FAM189A2 SORBS2 LRRN3 ABCA8 AOC3 CCM2L SFTPC ADRB1 TCF21 TGFBR3 HHIP ADH1B ARHGEF26 ZBTB16 ASPA FABP4 EDNRB SCN4B FCN3 ZBED2 MYCT1 KANK3 STX11 LINC00312 PLAC9 FAM107A CCDC85A CCBE1 AGER MARCO CD300LG TIE1 AGTR1 VIPR1 WIF1 RAMP3 CLIC5 FGFR4 FHL5 MAMDC2 CAMK2N1 AGTR2 CLDN18 C2orf40 CDH5 PDK4 GPM6A COL6A6 CFD GKN2 LRRC36 CYP4B1 HYAL1 TMEM100 DUOX1 AFF3 |
Table 2: Gene ontology analysis of all 162 common DEGs in NSCLC.
| Expression | Category | Term | Count | % | P-value | FDR |
|---|---|---|---|---|---|---|
| Up-regulated | GOTERM_BP_DIRECT | GO:0030574~collagen catabolic process | 7 | 17.07317 | 7.13E-09 | 9.86E-06 |
| GOTERM_BP_DIRECT | GO:0022617~extracellular matrix disassembly | 5 | 12.19512 | 2.83E-05 | 0.039054 | |
| GOTERM_BP_DIRECT | GO:0030199~collagen fibril organization | 4 | 9.756098 | 9.99E-05 | 0.138003 | |
| GOTERM_BP_DIRECT | GO:0007605~sensory perception of sound | 5 | 12.19512 | 2.50E-04 | 0.344372 | |
| GOTERM_BP_DIRECT | GO:0006508~proteolysis | 6 | 14.63415 | 0.005729 | 7.635062 | |
| GOTERM_CC_DIRECT | GO:0005578~proteinaceous extracellular matrix | 7 | 17.07317 | 2.08E-05 | 0.020878 | |
| GOTERM_CC_DIRECT | GO:0005581~collagen trimer | 5 | 12.19512 | 4.37E-05 | 0.043936 | |
| GOTERM_CC_DIRECT | GO:0005576~extracellular region | 12 | 29.26829 | 4.05E-04 | 0.406599 | |
| GOTERM_CC_DIRECT | GO:0005737~cytoplasm | 18 | 43.90244 | 0.03293 | 28.59139 | |
| Down-regulated | GOTERM_BP_DIRECT | GO:0001525~angiogenesis | 11 | 9.482759 | 5.81E-07 | 9.01E-04 |
| GOTERM_BP_DIRECT | GO:0001570~vasculogenesis | 5 | 4.310345 | 2.93E-04 | 0.454117 | |
| GOTERM_BP_DIRECT | GO:0007166~cell surface receptor signaling pathway | 8 | 6.896552 | 9.88E-04 | 1.520759 | |
| GOTERM_BP_DIRECT | GO:0031623~receptor internalization | 4 | 3.448276 | 0.001893 | 2.896712 | |
| GOTERM_BP_DIRECT | GO:0042310~vasoconstriction | 3 | 2.586207 | 0.00416 | 6.260617 | |
| GOTERM_CC_DIRECT | GO:0005887~integral component of plasma membrane | 24 | 20.68966 | 5.25E-06 | 0.006208 | |
| GOTERM_CC_DIRECT | GO:0016021~integral component of membrane | 49 | 42.24138 | 1.43E-04 | 0.169084 | |
| GOTERM_CC_DIRECT | GO:0045121~membrane raft | 8 | 6.896552 | 2.02E-04 | 0.237942 | |
| GOTERM_CC_DIRECT | GO:0005886~plasma membrane | 41 | 35.34483 | 3.06E-04 | 0.360544 | |
| GOTERM_CC_DIRECT | GO:0009897~external side of plasma membrane | 7 | 6.034483 | 0.001536 | 1.799873 | |
| GOTERM_MF_DIRECT | GO:0004872~receptor activity | 6 | 5.172414 | 0.006672 | 8.30861 | |
| GOTERM_MF_DIRECT | GO:0008201~heparin binding | 5 | 4.310345 | 0.011394 | 13.79895 | |
| GOTERM_MF_DIRECT | GO:0044325~ion channel binding | 4 | 3.448276 | 0.023849 | 26.85899 | |
| GOTERM_MF_DIRECT | GO:0005088~Ras guanyl-nucleotide exchange factor activity | 4 | 3.448276 | 0.024956 | 27.92675 | |
| GOTERM_MF_DIRECT | GO:0004945~angiotensin type II receptor activity | 2 | 1.724138 | 0.026957 | 29.82004 |
Table 3:KEGG pathway analysis of 162 common DEGs in NSCLC.
| Pathway ID | Pathway name | Count | % | P-value | Genes |
|---|---|---|---|---|---|
| hsa04512 | ECM-receptor interaction | 6 | 3.821656 | 0.001405 | CD36, COL6A6, COL1A1, COL11A1, SPP1,HMMR |
| hsa04514 | Cell adhesion molecules (CAMs) | 7 | 4.458599 | 0.002286 | CLDN18, PECAM1, CLDN5, JAM2, CDH3, SELE, CDH5 |
| hsa04670 | Leukocyte transendothelial migration | 6 | 3.821656 | 0.004752 | CLDN18, MMP9, PECAM1, CLDN5, JAM2, CDH5 |
| hsa04974 | Protein digestion and absorption | 5 | 3.184713 | 0.009863 | COL6A6, COL1A1, CPB2, COL11A1, COL10A1 |
| hsa03320 | PPAR signaling pathway | 4 | 2.547771 | 0.026133 | CD36, FABP4, ACADL, MMP1 |
| hsa04261 | Adrenergic signaling in cardiomyocytes | 5 | 3.184713 | 0.042961 | AGTR1, AGTR2, ADRB1, TNNC1, SCN4B |
| hsa04080 | Neuroactive ligand-receptor interaction | 7 | 4.458599 | 0.04882 | EDNRB, AGTR1, AGTR2, ADRB1, SSTR1, CALCRL, VIPR1 |
Analyzing of protein-protein interaction network (PPI) and modular
We applied the STRING database to build the PPI network, including 41 up-regulated genes and 121 down-regulated genes. A PPI network of the DEGs was presented as shown in figure 4a. Then we used Cytotype MCODE to construct a significant modular containing 10 nodes (ANLN, CCNA2, CDCA7, DEPDC1, DLGAP5, HMMR, KIAA0101, RRM2, TOP2A, and UBE2T) and 43 edges (Figure 4b). We discovered that these 10 central nodes were all up-regulated DEGs.
Figure 3
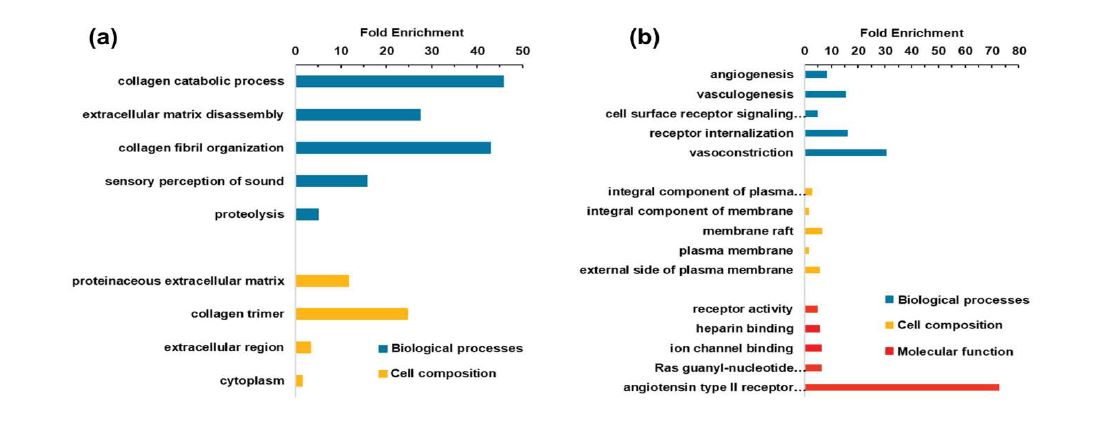
Figure 3: Gene ontology analysis of all 162 common DEGs in NSCLC from four gene expression datasets (GSE18842, GSE31210, GSE33532 and GSE101929) by using DAVID software. (a), GO enrichment analysis of 41 common up-regulated DEGs; (b), GO enrichment analysis of 121 common downregulated DEGs.
Figure 4
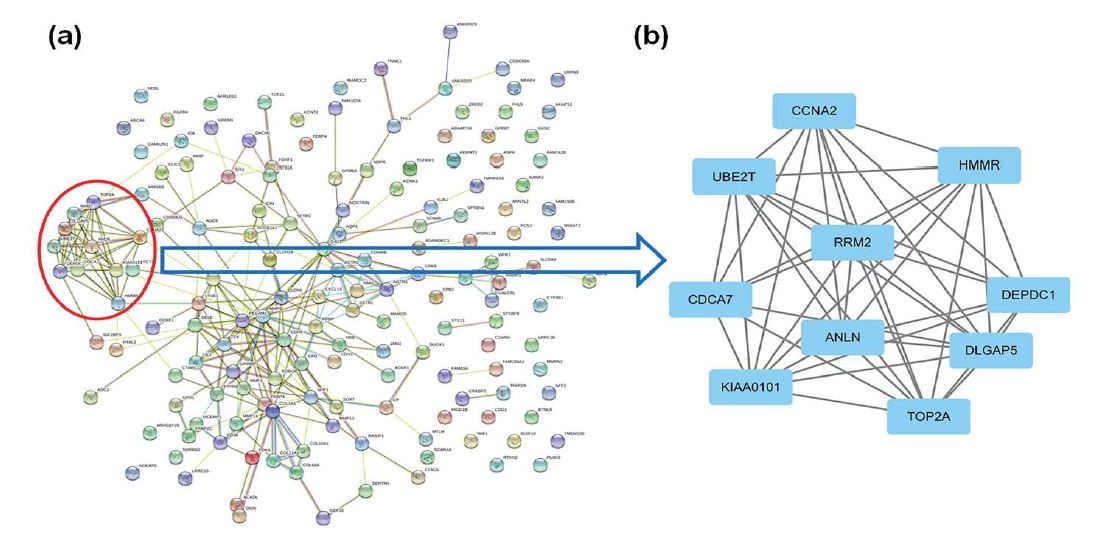
Figure 4:Constructing the PPI network of 162 common DEGs by using STRING database and analysis the modular through applying Cytotype MCODE. (a), The PPI network of 162 common DEGs in NSCLC; (b), A significant modular containing 10 nodes (ANLN, CCNA2, CDCA7, DEPDC1, DLGAP5, HMMR, KIAA0101, RRM2, TOP2A, and UBE2T) and 43 edges.
Analyzing of core genes
We next utilized Kaplan Meier-plotter and GEPIA to further analyze the 10 core genes. Kaplan Meier-plotter was used to illustrate the relationship between patients’ overall survival and gene expression levels of DEGs, while GEPIA to dig up the DEGs expression level between NSCLC and normal people. As shown in figure 5a, all the 10 core genes had an obviously worse survival when they had high expression in NSCLC patients (P<0.05). GEPIA results also demonstrated that all the 10 genes expressed higher in NSCLC samples than normal lung tissues, including LUAD and LUSC (P<0.05, Figure 5b).
Figure 5
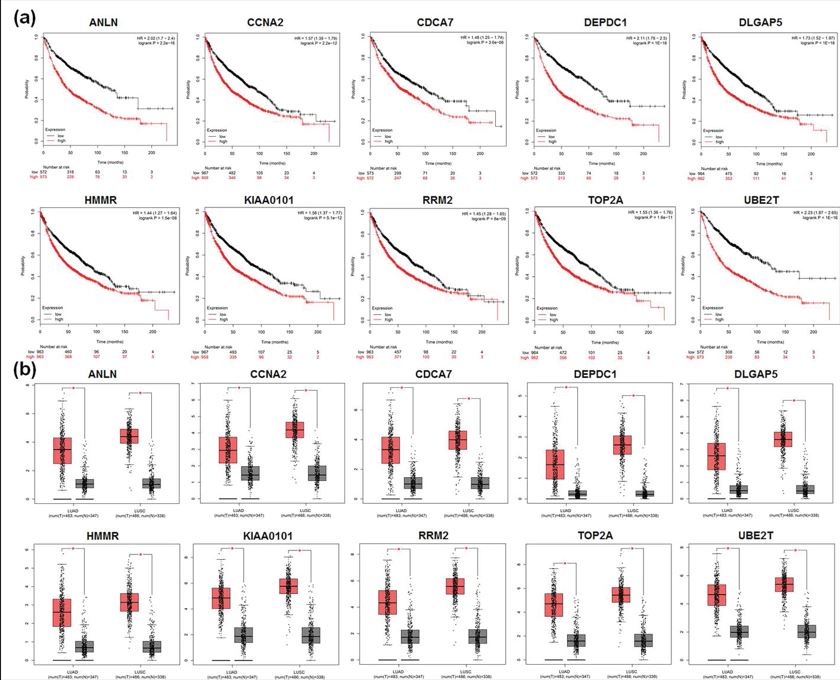
Figure 5: The important roles of the 10 core DEGs in NSCLC patients. (a), Analysis the relationship between NSCLC patients’ overall survival and gene expression levels of the 10 core DEGs by applying Kaplan Meier-plotter. As shown, all the 10 core genes had an obviously worse survival when they had high expression in NSCLC patients (P<0.05); (b), Analysis the 10 core DEGs expression level in NSCLC patients compared to healthy people by using GEPIA. As shown, all the 10 genes expressed higher in NSCLC samples than normal lung tissues, including LUAD and LUSC (*P<0.05). Red color means lung cancer tissues and grey color means normal lung tissues.
Discussion
As shown in figure 1, in this study, first, we together selected four NSCLC databases from GEO according to the screening principle which was described in the data source and preprocessing section; Second, we used GEO2R online tools to analyze the DEGs extracted from the four datasets respectively; Third, we applied Venn diagram software online to screen the common DEGs in these four datasets. In this part, we found that there were 162 DEGs in these four databases, including 41 up-regulated genes and 121 downregulated genes in NSCLC tissues; Fourth, we analyzed all the 162 DEGs GO enrichment and KEGG pathways by using DAVID software. As shown in table 3, the 162 DEGs were mainly enriched in ECM-receptor interaction, cell adhesion molecules, leukocyte transendothelial migration, protein digestion and absorption, PPAR signaling pathway, adrenergic signaling in cardiomyocytes and neuroactive ligand-receptor interaction to exert their biological function; Fifth, we constructed the PPI network of these 162 DEGs by applying the STRING database, then we discovered a significant modular containing 10 nodes through utilizing the Cytotype MCODE. These 10 core genes are ANLN, CCNA2, CDCA7, DEPDC1, DLGAP5, HMMR, KIAA0101, RRM2, TOP2A, and UBE2T; Last, we further analyzed the survival curve and the expression level between NSCLC tissues and normal lung tissues of these 10 core genes through Kaplan Meier plotter online database and GEPIA respectively. Taken together, we discovered that all the 10 genes were associated with poor prognosis in NSCLC, and they were all up-regulated DEGs.
ANLN (Anillin), an actin binding protein, is first found in Drosophila as a 124 kDa protein and plays an important role in cytokinesis [11]. ANLN has higher expression levels in the brain, testis, and placenta, but lower expression levels in the heart, kidney, liver, pancreas, prostate, spleen and lung. Recently, ANLN has been identified as a prognostic biomarker in cervical cancer, breast cancer, pancreatic cancer, colorectal cancer, and bladder urothelial carcinoma. ANLN is also discovered overexpressing in the majority of the primary NSCLC and is involved in the metastasis of lung cancer [12]. Pathway analysis demonstrated that ANLN participated in developmental processes through the regulation of nuclear division pathway [13].
CCNA2 (CyclinA2) belongs to a ubiquitously expressed member of the cyclin family and is expressed in almost all tissues in human [14]. Evidence indicated that CCNA2 was up-regulated in many kinds of cancers, and as an oncogenic gene, CCNA2 also played an important role in regulating cancer cell growth and apoptosis, especially controlling the cell cycle at the G1/S and the G2/M transitions [15]. CCNA2 can be used as a prognostic biomarker for colorectal cancer, ER+ breast cancer, esophageal squamous cell carcinoma and pancreatic etc. Resent study indicated that CCNA2 has higher expression in human NSCLC specimens than normal lung tissues, and could induce EMT and promote NSCLC metastasis via integrin αvβ3 signaling pathway [16]. However, further research is needed to uncover the target gene of CCNA2.
CDCA7 (Cell division cycle-associated protein 7), also known as JPO1, is a new member of cell division cycle associated genes family [17]. CDCA7 has been identified as a DNA-binding protein [18]. MYC and E2F1 could bind to the promoter of CDCA7, thereby driving CDCA7 expression. Recently, CDCA7 was discovered as a critical regulator of lymphomagenesis and invasion [19], while overexpression of CDCA7 predicted poor prognosis in triple negative breast cancer and colorectal cancer [20,21]. Wang’s study indicated that CDCA7 was significantly overexpressed in LUAD compared with the normal lung tissues, and silencing CDCA7 could inhibit cell proliferation through G1 phase arrest and induction of apoptosis [22]. In conclusion, CDCA7 can be considered as a therapeutic target for LUAD.
DEPDC1 (DEP domain containing 1), a highly conserved protein, plays important roles in many biological processes, for example, cell proliferation, cell cycle progression, cell apoptosis and signaling transduction etc [23]. DEPDC1 was firstly reported to be highly overexpressed in bladder cancer and had a critical role in the development of the bladder cancer [24]. Nowadays, DEPDC1 is considered as a novel oncoantigen which is upregulated in many kinds of cancers, including hepatocellular carcinoma, nasopharyngeal carcinoma, prostate cancer, breast cancer, and malignant glioma. DEPDC1 expression is also increased in LUAD and can be applied as a prognostic biomarker for NSCLC patients [25]. Recently, DEPDC1 was found inducing apoptosis in A549 lung adenocarcinoma cells by the NF-κB signaling pathway [26]. Further studies are needed to explore the mechanism of DEPDC1.
DLGAP5 (disc large homolog-associated protein 5), a mitotic spindle protein, can exert important biological function as a signaling molecule because it contains a guanylate-kinase-associated protein (GKAP) domain, which is highly conserved among many species and found in various eukaryotic signaling proteins [27] DLGAP5 overexpression could promote the proliferation potential of human cells, and the overexpression also been discovered in hepatocellular carcinoma, prostate cancer, colorectal cancer and adrenocortical carcinoma. Recently, studies also uncovered that DLGAP5 was highly overexpressed in the lung cancer tissues compared to corresponding normal lung tissues [28]. Hence, DLGAP5 can be used as promising biomarker for early detection of lung cancer.
DLGAP5 (disc large homolog-associated protein 5), a mitotic spindle protein, can exert important biological function as a signaling molecule because it contains a guanylate-kinase-associated protein (GKAP) domain, which is highly conserved among many species and found in various eukaryotic signaling proteins [27] DLGAP5 overexpression could promote the proliferation potential of human cells, and the overexpression also been discovered in hepatocellular carcinoma, prostate cancer, colorectal cancer and adrenocortical carcinoma. Recently, studies also uncovered that DLGAP5 was highly overexpressed in the lung cancer tissues compared to corresponding normal lung tissues [28]. Hence, DLGAP5 can be used as promising biomarker for early detection of lung cancer.
HMMR (Hyaluronan-mediated motility receptor), as an oncogene, is found highly up-regulated and plays important roles during the progression of human leukemias and solid tumors [29,30]. Tilghman’s work revealed that HMMR was overexpressed in glioblastoma (GBM) tumors where it supported the self-renewal and tumorigenic potential of GBM stem cells [31]. Taken together, HMMR not only promotes the progression of tumor, but also maintains the cancer stem cell (CSC) stemness. Meanwhile, some other studies have developed HMMR with great value for prognostic prediction in NSCLC [32]. But further research is needed to state the regulated mechanism of HMMR in NSCLC.
KIAA0101, also named as proliferating cell nuclear antigen (PCNA)-associated factor (PAF15), functions as an oncogene and is upregulated in various cancers, including breast cancer, esophageal cancer, hepatocellular carcinoma, ovarian cancer and lung cancer. KIAA0101 has been recently considered as a potential biomarker for recurrence and poor prognosis in tumor patients. Kato’s study discovered that KIAA0101 was overexpressed in the great majority of lung cancers, and KIAA0101 could be used as a specific target to treat lung cancer [33].
RRM2 (Ribonucleotide reductase M2 subunit), a small subunit of the ribonucleotide reductase complex, is a ratelimiting enzyme for dNTP producing and displays critical roles in many cellular processes such as cell proliferation, invasiveness, migration and angiogenesis [34]. RRM2 has been reported overexpressing in various malignancies as a tumor driver, including breast cancer, gliomas, colorectal cancer, bladder cancer and NSCLC. Yang’s work found RRM2 was upregulated in NSCLC tumor and cell lines, and the aberrant upregulation predicted a poor prognosis [35]. Mechanistically, they also revealed the vital role of LINC00667/miR-143-3p/RRM2 signal pathway in the NSCLC progression. In conclusion, RRM2 can be used as a therapeutic target for NSCLC.
TOP2A (Topoisomerase 2-alpha) encodes a nuclear enzyme which implicates in almost any processes of DNA metabolism, such as replication, transcription and chromosome segregation during interphase and mitosis [36]. It has been reported that TOP2A has higher expression level in a variety of human cancers, including gastric cancer, bladder urothelial carcinoma, colon cancer and pancreatic cancer. Meanwhile, TOP2A also can be considered as the target for some of the most widely used chemotherapeutic drugs for human cancers treatment [37]. But the role of TOP2A in progression of NSCLC has not been elucidated.
UBE2T (Ubiquitin-conjugating enzyme E2T, also named as HSPC150), a member of the E2 family, is firstly identified in a patient with Fanconi anemia (FA) [38]. UBE2T takes part in main cellular processes such as cell cycle control, signal transduction and tumorigenesis through working with specific E3 ubiquitin ligase to active the degradation of relevant substrates [39]. UBE2T has been also discovered overexpressed in prostate cancer, osteosarcoma, gastric cancer, hepatocellular carcinoma and lung cancer. But the mechanism of UBE2T to promote the progression of NSCLC is not clear now. Further studies are needed to clarify the relationship between UBE2T and NSCLC.
Conclusion
In conclusion, we discovered 10 key oncogenes which were associated with the progression and poor prognosis for NSCLC through our research, including ANLN, CCNA2, CDCA7, DEPDC1, DLGAP5, HMMR, KIAA0101, RRM2, TOP2A, and UBE2T. These 10 genes can be served as the therapeutic targets and useful prognostic biomarkers for NSCLC treatment. But the mechanism of these genes to regulate the progression of NSCLC is needed to explore, it is useful to design new drugs targeting these oncogenes.
Statement of Ethics
This article does not contain any studies with human participants or animals performed by any of the authors.
Declaration of Conflicting Interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and publication of this article.
Author Contributions
L Wang and N Hu conceived and designed the idea to this manuscript; W Wu and C Fang collected and analyzed the data, and drafted the manuscript; C Zhang collected the data and revised the manuscript. All authors confirmed the final version of the manuscript for submission.
Funding
This work was supported by National Natural Science Foundation of China (Grant No. 81803933) and Xinglin Young Talent Program of Shanghai University of Traditional Chinese Medicine (Grant No. A1-R20-409-01-0301).

View pdf
Download pdf
Miller KD, Goding Sauer A, Ortiz AP, Fedewa SA, Pinheiro PS, et al. (2018) Cancer Statistics for Hispanics/Latinos, 2018. CA Cancer J Clin 68: 425-445. [ Ref ]
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, et al. (2013) Cancer genome landscapes. Science 339: 1546-1558. [ Ref ]
Falzone L, Lupo G, La Rosa GRM, Crimi S, Anfuso CD, et al. (2019) Identification of Novel MicroRNAs and Their Diagnostic and Prognostic Significance in Oral Cancer. Cancers 11: 610. [ Ref ]
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4: 44-57. [ Ref ]
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene Ontology: tool for the unification of biology. Nature Genetics 25: 25-29. [ Ref ]
Zhong M, Wu YL, Ou WJ, Huang LJ, Yang LY (2019) Identification of key genes involved in type 2 diabetic islet dysfunction: a bioinformatics study. Bioscience Reports 39: Bsr20182172. [ Ref ]
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498-2504. [ Ref ]
Feng H, Gu ZY, Li Q, Liu QH, Yang XY, et al. (2019) Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J Ovarian Res 12: 35. [ Ref ]
- Szasz AM, Lanczky A, Nagy A, Forster S, Hark K, et al. (2016) Crossvalidation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 7: 49322-49333.
Tang Z, Li C, Kang B, Gao G, Li C, et al. (2017) GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45(W1): W98-W102. [ Ref ]
Piekny AJ, Maddox AS (2010) The myriad roles of Anillin during cytokinesis. Semin Cell Dev Biol 21: 881-891. [ Ref ]
Xu J, Zheng H, Yuan S, Zhou B, Zhao, W, et al. (2019) Overexpression of ANLN in lung adenocarcinoma is associated with metastasis. Thorac Cancer 10: 1702-1709. [ Ref ]
Long X, Zhou W, Wang Y, Liu S (2018) Prognostic significance of ANLN in lung adenocarcinoma. Oncol Lett 16: 1835-1840. [ Ref ]
Ko E, Kim Y, Cho EY, Han J, Shim YM, et al. (2013) Synergistic effect of Bcl-2 and cyclin A2 on adverse recurrence-free survival in stage I nonsmall cell lung cancer. Ann Surg Oncol 20: 1005-1012. [ Ref ]
Gan Y, Li Y, Li T, Shu G, Yin G (2018) CCNA2 acts as a novel biomarker in regulating the growth and apoptosis of colorectal cancer. Cancer Manag Res 10: 5113-5124. [ Ref ]
Ruan JS, Zhou H, Yang L, Wang L, Jiang ZS, et al. (2017) CCNA2 facilitates epithelial-to-mesenchymal transition via the integrin alphavbeta3 signaling in NSCLC. Int J Clin Exp Pathol 10: 8324-8333. [ Ref ]
Guiu J, Bergen DJ, De Pater E, Islam AB, Ayllon V, et al. (2014) Identification of Cdca7 as a novel Notch transcriptional target involved in hematopoietic stem cell emergence. J Exp Med 211: 2411-2423. [ Ref ]
Prescott JE, Osthus RC, Lee LA, Lewis BC, Shim H, et al. (2001) A novel c-Myc-responsive gene, JPO1, participates in neoplastic transformation. J Biol Chem 276: 48276-48284. [ Ref ]
Jimenez PR, Martin-Cortazar C, Kourani O, Chiodo Y, Cordoba R, et al. (2018) CDCA7 is a critical mediator of lymphomagenesis that selectively regulates anchorage-independent growth. Haematologica, 103: 1669- 1678. [ Ref ]
Li D, Jiang X, Zhang X, Cao G, Wang D, et al. (2019) Long noncoding RNA FGD5-AS1 promotes colorectal cancer cell proliferation, migration, and invasion through upregulating CDCA7 via sponging miR-302e. In Vitro Cell Dev Biol Anim 55: 577-585. [ Ref ]
Ye L, Li F, Song Y, Yu D, Xiong Z, et al. (2018) Overexpression of CDCA7 predicts poor prognosis and induces EZH2-mediated progression of triple-negative breast cancer. Int J Cancer 143: 2602-2613. [ Ref ]
Wang H, Ye L, Xing Z, Li H, Lv T, et al. (2019) CDCA7 promotes lung adenocarcinoma proliferation via regulating the cell cycle. Pathol Res Pract 215: 152559. [ Ref ]
Zhou C, Wang P, Tu M, Huang Y, Xiong F, et al. (2019) DEPDC1 promotes cell proliferation and suppresses sensitivity to chemotherapy in human hepatocellular carcinoma. Biosci Rep 39: BSR20190946. [ Ref ]
Kanehira M, Harada Y, Takata R, Shuin T, Miki T, et al. (2007) Involvement of upregulation of DEPDC1 (DEP domain containing 1) in bladder carcinogenesis. Oncogene 26: 6448-6455. [ Ref ]
Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, et al. (2012) Identification of genes upregulated in ALK-positive and EGFR/KRAS/ ALK-negative lung adenocarcinomas. Cancer Res 72: 100-111. [ Ref ]
Wang Q, Li A, Jin J, Huang G (2017) Targeted interfering DEP domain containing 1 protein induces apoptosis in A549 lung adenocarcinoma cells through the NF-kappaB signaling pathway. Onco Targets Ther 10: 4443-4454. [ Ref ]
Liao W, Liu W, Yuan Q, Liu X, Ou Y, et al. (2013) Silencing of DLGAP5 by siRNA significantly inhibits the proliferation and invasion of hepatocellular carcinoma cells. Plos One 8: e80789. [ Ref ]
- Schneider MA, Christopoulos P, Muley T, Warth A, Klingmueller U, et al. (2017) AURKA, DLGAP5, TPX2, KIF11 and CKAP5: Five specific mitosisassociated genes correlate with poor prognosis for non-small cell lung cancer patients. Int J Oncol 50: 365-372.
Giannopoulos K, Li L, Bojarska-Junak A, Rolinski J, Dmoszynska A, et al. (2006) Expression of RHAMM/CD168 and other tumor-associated antigens in patients with B-cell chronic lymphocytic leukemia. Int J Oncol 29: 95-103. [ Ref ]
Maxwell CA, McCarthy J, Turley E (2008) Cell-surface and mitoticspindle RHAMM: moonlighting or dual oncogenic functions? J Cell Sci 121: 925-932. [ Ref ]
Tilghman J, Wu H, Sang Y, Shi X, Guerrero-Cazares H, et al. (2014) HMMR maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer Res, 74: 3168-3179. [ Ref ]
He R, Zuo S (2019) A Robust 8-Gene Prognostic Signature for Early-Stage Non-small Cell Lung Cancer. Front Oncol 9: 693. [ Ref ]
Kato T, Daigo Y, Aragaki M, Ishikawa K, Sato M, et al. (2012) Overexpression of KIAA0101 predicts poor prognosis in primary lung cancer patients. Lung Cancer 75: 110-118. [ Ref ]
Nordlund P, Reichard P (2006) Ribonucleotide reductases. Annu Rev Biochem 75: 681-706. [ Ref ]
Yang Y, Li S, Cao J, Li Y, Hu H, et al. (2019) RRM2 Regulated By LINC00667/miR-143-3p Signal Is Responsible For Non-Small Cell Lung Cancer Cell Progression. Onco Targets Ther 12: 9927-9939. [ Ref ]
Nuncia-Cantarero M, Martinez-Canales S, Andres-Pretel F, Santpere G, Ocana A, et al. (2018) Functional transcriptomic annotation and proteinprotein interaction network analysis identify NEK2, BIRC5, and TOP2A as potential targets in obese patients with luminal A breast cancer. Breast Cancer Res Treat 168: 613-623. [ Ref ]
Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9: 338-350. [ Ref ]
Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM,et al. (2006) UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell 23: 589-596. [ Ref ]
Lim KH, Song MH, Baek KH (2016) Decision for cell fate: deubiquitinating enzymes in cell cycle checkpoint. Cell Mol Life Sci 73: 1439-1455. [ Ref ]
