Journal Name: Journal of Biomedical Research and Reviews
Article Type: Research
Received date: 21 February, 2020
Accepted date: 11 March, 2020
Published date: 2021-03-21
Citation: Lee M (2020) Investigating the Influence of the Comprehensive mRNA Expression Levels of Prognostic Genes on Patient Survival in Every Type of Cancer. J Biomed Res Rev Vol: 3, Issu: 1 (18-21).
Copyright: © 2020 Lee M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
This study aimed to rank cancers based on the strength of the relationship between the comprehensive mRNA expression levels of the most harmful or protective genes and patient survival. Using The Cancer Genome Atlas dataset that includes the RNA sequencing and clinical data, we investigated not only gene specific prognostic availability, but also comprehensive prognostic availability of prognostic genes filtered by the Cox coefficient values, and ranked cancers using a specially designed prognostic indicator. Using Kaplan–Meier plots, we found that cancers vary in the strength of the influence of their prognostic genes, and can be ranked based on this finding. There is a high probability that the treatment developed by using methods that reduce or increase the expression levels of biomarkers, for cancers that ranked at the bottom will not be efficient. The results of this study could be used as scientific evidence for the same.
KeyWords
Bioinformatics, Genomics, TCGA, Cox Model.
Abstract
This study aimed to rank cancers based on the strength of the relationship between the comprehensive mRNA expression levels of the most harmful or protective genes and patient survival. Using The Cancer Genome Atlas dataset that includes the RNA sequencing and clinical data, we investigated not only gene specific prognostic availability, but also comprehensive prognostic availability of prognostic genes filtered by the Cox coefficient values, and ranked cancers using a specially designed prognostic indicator. Using Kaplan–Meier plots, we found that cancers vary in the strength of the influence of their prognostic genes, and can be ranked based on this finding. There is a high probability that the treatment developed by using methods that reduce or increase the expression levels of biomarkers, for cancers that ranked at the bottom will not be efficient. The results of this study could be used as scientific evidence for the same.
KeyWords
Bioinformatics, Genomics, TCGA, Cox Model.
Introduction
To perform the most comprehensive research possible, we used the Cancer Genome Atlas (TCGA) dataset including clinical, whole genome sequencing, exome sequencing, RNA sequencing (RNA-Seq), small RNASeq, bisulfite-Seq, and reverse phase array information to identify the pathways commonly altered in different cancers [1-11].
There are various approaches for cancer therapy, such as surgery, radiotherapy, immunotherapy, and chemotherapy. Usually, most of the chemotherapeutic methods reduce or increase the expression levels of the proteins or genes related to patient survival. However, few biomarkers can be used efficiently in the treatment of some cancers. This research provided scientific evidence of the significant relationships between the clinical outcomes and the comprehensive mRNA expression levels of prognostic genes in some types of cancer. In this study, we formulated an indicator called the prognostic score. The results of this study can be used as a guide in the development of the cure for some cancers.
Materials and Methods
Codes and files
All files and R codes generated for this study, including figures and tables, can be downloaded from the author’s github page (https://github.com/Minhyeong-1022/TCGA_mRNA-expression_survival_ correlation_research). The scripts were run on a Samsung laptop 9 with an i5 processor and 8 GB RAM, running R 3.5.1 on Windows 10.
Construction of the prognostic indicator model
To convert the RNA-Seq and clinical data into R source codes, we used the getFirehoseData function from the R library named RTCGAToolbox, which pulls TCGA data from https://tcga-data.nci.nih.gov/tcga/, in September 2019. All clinical follow-up information was extracted from the “clinical” files for each cancer, and only patient information of the clinical events was included in this analysis. TCGA dataset provides mRNA expression data obtained from two methods, RNA-Seq and RNA-SeqV2. We used RNA-SeqV2 because it is the most recent version. “RNASeq2GeneNorm” files were formed by RSEM, which is a method of reporting the mRNA expression levels, and outputted by the RSEM software [12]. The genes, for each cancer, with expression values greater than 1 were included in this model. The prognostic scores were determined from squared regression (R2) values, and the ratio of genes with p-values less than or equal to 0.01 was generated from each term of the Cox model. The R2 values, which are the Cox coefficient and Pearson correlation coefficient values of R1 values, were generated for each cancer. The R1 values, which are the Pearson correlation coefficient values of patient-specific survival days to death and mRNA expression values (RSEM) of each gene, were generated for each gene. The information from TCGA dataset, including the number of genes selected using the p-values generated from the Cox model, are represented in Table 1. Prognostic scores were also generated for each cancer. The formula used to generate the comprehensive prognostic indicator is represented below.
R1 = Pearson cor (mRNA expression values, Days to death)
R2 = Pearson cor (Cox coefficient, R1)
Table 1: Characteristics of TCGA dataset and investigated numbers of selected genes.
| Cancer type | Number of patients | Number of total genes | Number of selected genes (Raw P-value < 0.01) |
|---|---|---|---|
| OV | 591 | 16923 | 115 |
| BRCA | 1097 | 16632 | 695 |
| COAD | 458 | 16408 | 963 |
| BLCA | 412 | 16367 | 1557 |
| KIRP | 291 | 16429 | 3305 |
| CESC | 307 | 16359 | 951 |
| LUAD | 522 | 16777 | 2028 |
| STAD | 443 | 16914 | 852 |
| HNSC | 528 | 16642 | 832 |
| LUSC | 504 | 16972 | 180 |
| LAML | 200 | 15251 | 599 |
| SKCM | 470 | 16058 | 2400 |
| LIHC | 377 | 15850 | 1766 |
| KIRC | 537 | 16665 | 5447 |
Information of the multivariate Cox model used in the analysis
Gene-specific Cox coefficient values and p-values for 16 cancers were generated in the previous study, published in February 2016, by Jordan Anaya at University of Virginia [13,14]. We used the data file “All genes, p-values, Cox coefficients for each cancer,” and it can be downloaded online at (https://peerj.com/articles/1499/#supp-1).
Results
We analyzed TCGA data on a number of patients with RNA-Seq data and mature clinical follow-up information. Fourteen cancers, including acute myeloid leukemia (AML), bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), colon adenocarcinoma (COAD), head and neck squamous cell carcinoma (HNSC), kidney clear cell renal carcinoma (KIRC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), ovarian serous cystadenocarcinoma (OV), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), kidney papillary renal cell carcinoma (KIRP), liver hepatocellular carcinoma (LIHC), skin cutaneous melanoma (SKCM), and stomach adenocarcinoma (STAD) were studied.
This study aimed at ranking cancers, based on the sensitivity of the cancer to the expression levels of their prognostic genes, using a specially formulated indicator called the prognostic score. To achieve this, the multivariate Cox proportions hazards model [15], which is a standard regression method to research clinical data [16-19], was used. The data table in the previous study [13,14], including gene-specific Cox coefficient values and p-values for 16 cancers, was used.
In previous studies, the Cox coefficient values and p-values, provided from each term of the Cox model, were used to analyze the influence of the mRNA expression level of each gene on patient survival. There were significant deviations of patient survival patterns, based on the expression levels of genes that have extreme Cox coefficient values, and we checked these deviations using Kaplan– Meier plots for all cancers (Figure 1d-g). Patient survival in some types of cancers was significantly influenced by the expression levels of the genes. This was however, not the case for other types of cancers because cancers vary in their sensitivity to the expression levels of prognostic genes. We calculated the prognostic scores for all types of cancers in this study. Patient survival was influenced by not only the R2 values, but also the ratio of the selected genes. The cancers differed in their prognostic scores, and a bar chart showing the ranking of the cancers is displayed in Figure 1. The expression levels of the prognostic genes of the top ranked cancers influenced the patient clinical outcomes more than the expression levels of those of the bottom ranked cancers.
Figure 1
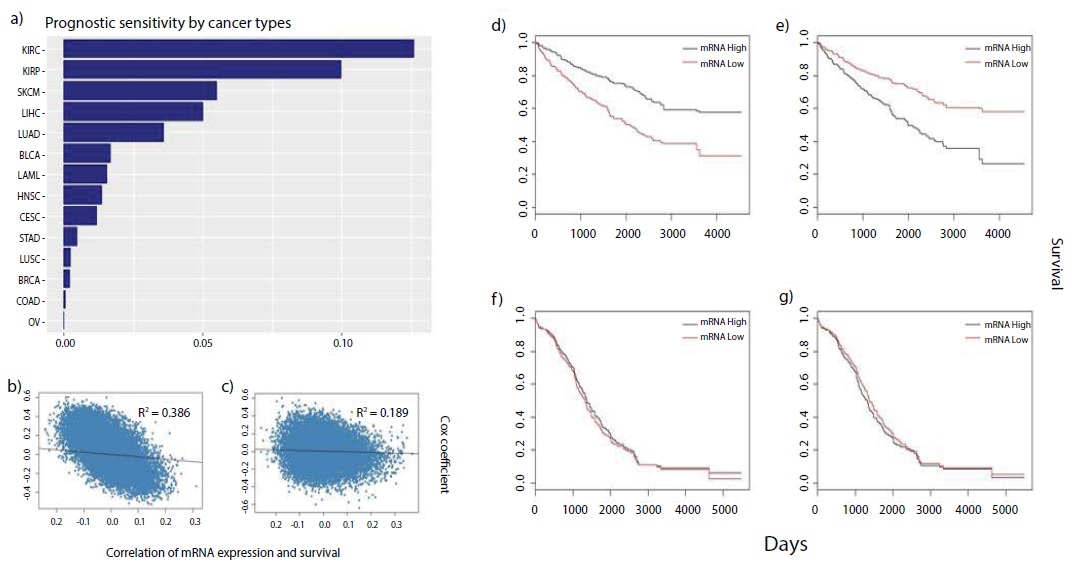
Figure 1: Distinct difference of survival patterns by prognostic score.
(a) Bar-chart shows the rank of cancers by prognostic score. Prognostic score was mostly determined by R2 which is the squared regression of cox coefficient
and the Pearson correlation coefficient (R1) of patient-specific survival days to death and mRNA expression for each gene. (b) Scatter plot related to R2 of
KIRC which ranked top in bar-chart and (c) R2 of OV which ranked bottom in bar-chart. (d), (e), (f), (g) Kaplan-Meier plots comparing survival days of KIRC
to survival days of OV patients for the 100 most protective genes and for the 100 most harmful genes.
For all of 14 cancers used in this study, the numbers of total genes and the numbers of genes selected by raw p-value of less than or equal to 0.01 were represented on the table above. The ratio of these two would be factor of prognostic indicator.
The R2 value of KIRC, which had the highest prognostic score among the 14 cancers, (Figure 1b) was more significant than that of OV (Figure 1c), which was ranked at the bottom. KIRC and OV markedly differed in their patient survival patterns, obtained using the expression levels of their prognostic genes. As shown in figure 1, KIRC, unlike OV, was found to be very sensitive to gene expression. The prognostic sensitivity of the other cancers to the expression levels of their prognostic genes followed the ranking (Figure 1a). Supplemental materials, including Kaplan–Meier plots and scatter plots for other cancers, can be downloaded from the github page.
Discussion
The mRNA expression levels of protective or harmful genes, filtered by the Cox coefficient values, influence patient survival differently in different types of cancers. To rank the cancers, based on the influence of the expression levels of the prognostic genes on clinical outcomes, a specially formulated indicator called the prognostic score was used in this study. In conclusion, it was successfully used to predict the ranking of the different types of cancers. The results of this study can be used as scientific evidence for the development of the cure for different types of cancer by considering the sensitivity of the cancer to the expression levels of its prognostic genes. For example, as shown in figure 1, there is a high probability that the treatment developed for OV with chemotherapeutic methods that reduce or increase the mRNA expression and protein levels will not be efficient. However, developing treatment for KIRC with chemotherapeutic methods could be successful.
Conflict of Interest
No potential conflict of interest relevant to this article is reported.

View pdf
Download pdf
There are no references
