Journal Name: Journal of Biomedical Research and Reviews
Article Type: Mini Review
Received date: 20 August, 2018
Accepted date: 21 August, 2018
Published date: 27 August, 2018
Citation: Yin CY, Manap ASBA (2018) The Overview of Alzheimer Disease Intricate Mechanism: Amyloidopathy and its Toxicity. J Biomed Res Rev Vol: 1, Issu: 2 (23-24).
Copyright: © 2018 Yin CY. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Alzheimer’s disease (AD) is the most widely recognised neurodegenerative disorder, characterised by progressive neuronal loss with major deposition of amyloid β-peptide (Aβ) plaques. Amyloidopathy is the key element in the pathogenesis of AD that pathologically induced massive neuronal atrophy and damage of synapses in different brain regions that leading to neuronal death and memory loss. Aβ peptide derived from amyloid precursor protein (APP) in amyloidogenic pathway is deposited as plaques in the brain, which are said to be the hallmark of Alzheimer’s disease. Moreover, excess Aβ production, its aggregation and deposition deleteriously affect a large number of biologically important pathways leading to neuronal cell death. This mini review intends to detail in the mechanism of amyloidopathy of Aβ peptide and emphasize its toxic effects that play a crucial role in the pathogenesis of AD. Perhaps in future, these are the areas in which investigation on the modulation of Aβ accumulation in the prevention of AD could be focused on.
Keywords
Alzheimer’s disease, Amyloidopathy, Amyloid beta, Amyloid precursor protein.
Abstract
Alzheimer’s disease (AD) is the most widely recognised neurodegenerative disorder, characterised by progressive neuronal loss with major deposition of amyloid β-peptide (Aβ) plaques. Amyloidopathy is the key element in the pathogenesis of AD that pathologically induced massive neuronal atrophy and damage of synapses in different brain regions that leading to neuronal death and memory loss. Aβ peptide derived from amyloid precursor protein (APP) in amyloidogenic pathway is deposited as plaques in the brain, which are said to be the hallmark of Alzheimer’s disease. Moreover, excess Aβ production, its aggregation and deposition deleteriously affect a large number of biologically important pathways leading to neuronal cell death. This mini review intends to detail in the mechanism of amyloidopathy of Aβ peptide and emphasize its toxic effects that play a crucial role in the pathogenesis of AD. Perhaps in future, these are the areas in which investigation on the modulation of Aβ accumulation in the prevention of AD could be focused on.
Keywords
Alzheimer’s disease, Amyloidopathy, Amyloid beta, Amyloid precursor protein.
Introduction
Alzheimer disease (AD) is the most devastating, progressive and irreversible neurodegenerative disorder, which primarily affects the elderly [1]. This disease is the most common and feared type of dementia representing circa 70% of all dementia cases and displaying dramatic epidemics worldwide. The AD is mainly characterized by the deterioration of both the memory and cognitive ability [2]. Likewise, other debilitating non-cognitive manifestations arise as the disease progress, such as visual hallucination, depression, impaired sleep as well appetite [3,4].
“Amyloidopathy” refers to the accumulation of amyloid beta (Aβ) peptide, which is the core element of senile plaques in neuronal cells. It is pictured that the extracellular aggregation of Aβ plaques is closely related with extensive neuronal atrophy and the contemporaneous destruction of synapses in different brain regions that consequences in gradual neuronal death and memory impairment. A study by Hardy [5] has documented that several environmental factors, including oxidative stress, drugs, brain injury, and genetic divergences, capable to contribute to the pathogenesis of AD. The amyloid cascade hypothesis has well portrayed the mechanism of deposition of the Aβ protein in Alzheimer’s pathology [6].
Pathogenesis of Amyloid Beta (Aβ)
Amyloid precursor protein (APP) is a large transmembrane protein that generates Aβ that comprises of either 40 or 42 amino acids. Aβ40 is a normally found protein molecule whereby Aβ42 is highly toxic to the neuronal cells. In physiological states, the membrane glycoprotein APP plays a crucial role in synapse development, neuronal transmission and activity via two proteolytic pathways: “nonamyloidogenic” and “amyloidogenic” pathways [7]. In the “nonamyloidogenic” pathway, soluble APP𝛼 is released to the extracellular space through APP cleavage that involves 𝛼- and β-secretases. While, the “amyloidogenic” pathway is triggered by β-secretase 1 (β-site amyloid precursor protein cleaving enzyme 1, BACE-1) mediated cleavage that forms soluble extracellular APPβ. Next, this APPβ was cleaved by a 𝛾-secretase complex that emitted Aβ into the extracellular compartment [8]. Subsequently, Aβ is transported and degraded by the Apolipoprotein (Apo) E2 and ApoE3 isoform via the blood brain barrier (BBB) together with insulin degrading enzyme (IDE) or neprilysin degradation pathway (NDP). However, the binding of Aβ to the ApoE4 isoform cause major Aβ aggregation in the extracellular region of the central nervous system. In normal circumstances, during Aβ generation, there is a feedback mechanism that releases the APP intracellular C-terminal domain and raises the level of neprilysin, which promotes Aβ turnover. Disruptions of the normal physiological mechanism may occur in the event of mutations and changes in the expression of APP, BACE-1, IDE, Apo-E, and neprilysin leads to the progressive accretion of Aβ resulting to the manifestation of AD [9].
The amyloid beta hypothesis proposes that overload Aβ production, aggregation and its accumulation in the brain as plaques is the key reason for the progression of AD. Although Aβ is considered toxic, there is always a certain level of Aβ peptide (picomolar) sustained in the brain and CSF supporting the fact that Aβ has a positive effect on diverse regulatory aspects of physiological function of neuronal. Aβ accumulations are considered to play a crucial role in the pathogenesis of AD by inducing oxidative stress, synaptic dysfunction, cell membrane disruption, mitochondria dysregulation and apoptosis (Figure 1). It is however, the exact mechanism of toxicity induced by Aβ aggregates is still a matter of debate. Several hypotheses have been proposed in current literature all of which concur that in some way or the other Aβ aggregation plays a prominent role in AD. Therefore, the modulation of Aβ aggregation such as using peptides and small molecule-based inhibitors is a promising approach [10,11].
Figure 1
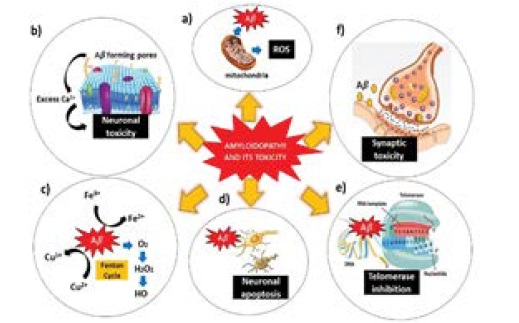
Figure 1: Amyloidopathy and its toxicity. a) Aβ cause mitochondrial dysfunction leading to ROS generation. b) Cell membrane disruption by Aβ aggregates. c) Oxidative stress caused by Aβ oligomers. (d) Aβ induce neuronal apoptosis. e) Telomerase inhibition. f) Aβ interfere with signaling pathways causing synaptic toxicity [Adaptation and Modifications from Rajasekhar et al. [10]].
Conclusion
The critical role of Aβ aggregation and deposition in AD has generated colossal interest among the scientist to understand its role in neurodegeneration. In this mini review, we have highlighted mechanism of Aβ deposition and toxicity induced by its aggregates. Aβ is produced both by amyloidogenic and non-amyloidogenenic pathways by APP. Pathological hallmark of AD is characterised by amyloidogenenic pathway that involved massive production of toxic variants of Aβ which consequently affects various biological pathways in AD. This short review has provided valuable insight of Aβ mechanism and its significance as a therapeutic target in AD.
Acknowledgement
Authors are thankful to Taylor’s University Malaysia for their support and facilities provided, Department of Health and Sciences and Ministry of Higher Education, Malaysia.
Conflict of Interest
All the authors ensure that there is no conflict of interest regarding authorship or any other matters pertaining to this manuscript.

View pdf
Download pdf
Singh A (2010) Medicinal plant for curing Alzheimer’s disease. International Journal of Pharmaceutical and Biological Archive. [ Ref ]
Jadoopat R (2018) Review of Alzheimer’s Disease Treatment and Potential Future Therapies. Annual Review of Changes in Healthcare 2: 1–10. [ Ref ]
Ishii M, Iadecola C (2015) Metabolic and non-cognitive manifestations of Alzheimer’s disease: the hypothalamus as both culprit and target of pathology. Cell metabolism 22: 761–776. [ Ref ]
Lanctôt KL, Amatniek J, Ancoli-Israel S, Arnold SE, Ballard C, et al. (2017) Neuropsychiatric signs and symptoms of Alzheimer’s disease: new treatment paradigms. Alzheimer’s and Dementia: Translational Research & Clinical Interventions 3: 440–449. [ Ref ]
Hardy J (2006) Alzheimer’s disease: the amyloid cascade hypothesis: an update and reappraisal. Journal of Alzheimer’s disease 9: 151–153. [ Ref ]
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256: 184–185. [ Ref ]
Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, et al. (2006) Synapse formation and function is modulated by the amyloid precursor protein. Journal of Neuroscience 26: 7212–7221. [ Ref ]
Ma H, Lesné S, Kotilinek L, Steidl-Nichols JV, Sherman M, Younkin L, et al. (2007) Involvement of β-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activitydependent synaptic plasticity. Proceedings of the National Academy of Sciences 104: 8167–8172. [ Ref ]
Kim J, Basak JM, Holtzman DM (2009) The role of apolipoprotein E in Alzheimer’s disease. Neuron 63: 287–303. [ Ref ]
Rajasekhar K, Chakrabarti M, Govindaraju T (2015) Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chemical Communications 51: 13434–13450. [ Ref ]
Hayden EY, Teplow DB (2013) Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimer’s research & therapy 5: 60. [ Ref ]
