Journal Name: Journal of Clinical Case Reports and Trials
Article Type: Case Report
Received date: 31 March, 2021
Accepted date: 10 April, 2021
Published date: 12 April, 2021
Citation: Condorelli A, Markovic U, Nicolosi D, Calafiore V, Giorgio MAD et al. (2021) Gaucher Disease Type 1 In A 4-Year Old Child With Igg-K Mgus Successfully Treated With Imiglucerase: A Case Report. J Clin Case Rep Trials. Vol: 4, Issu: 1 (11-14).
Copyright: © 2021 Condorelli A et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The association between monoclonal gammopathy of undetermined significance (MGUS), Multiple Myeloma (MM) and Gaucher disease (GD) is well described in the literature. The risk for MM in GD patients may be 25- to 50-fold greater than expected in the general population. The probable pathophysiology is the acquisition of somatic mutations secondary to the chronic immune stimulation driven by the augmented glycosilceramide or its deacylated products in the macrophages. Here we described the case of a 4-year old patient diagnosed with Gaucher disease with GD-related IgG-k MGUS in the setting of a prospective observational study on the prevalence of underrecognized type I Gaucher disease in a selected population with MGUS. The detection of MGUS, together with anemia and splenomegaly, allowed us to achieve an early diagnosis and to immediately start enzymatic replacement treatment (ERT). The prompt start of ERT resulted in the prevention of long- term complications of the disease and in the disappearance of the monoclonal protein, thus likely reducing the risk of MM.
Keywords
Gaucher Disease; Monoclonal Gammopath; Multiple Myeloma; Enzymatic Replacement Treatment
Abstract
The association between monoclonal gammopathy of undetermined significance (MGUS), Multiple Myeloma (MM) and Gaucher disease (GD) is well described in the literature. The risk for MM in GD patients may be 25- to 50-fold greater than expected in the general population. The probable pathophysiology is the acquisition of somatic mutations secondary to the chronic immune stimulation driven by the augmented glycosilceramide or its deacylated products in the macrophages. Here we described the case of a 4-year old patient diagnosed with Gaucher disease with GD-related IgG-k MGUS in the setting of a prospective observational study on the prevalence of underrecognized type I Gaucher disease in a selected population with MGUS. The detection of MGUS, together with anemia and splenomegaly, allowed us to achieve an early diagnosis and to immediately start enzymatic replacement treatment (ERT). The prompt start of ERT resulted in the prevention of long- term complications of the disease and in the disappearance of the monoclonal protein, thus likely reducing the risk of MM.
Keywords
Gaucher Disease; Monoclonal Gammopath; Multiple Myeloma; Enzymatic Replacement Treatment
Introduction
Gaucher disease (GD) is an autosomal recessive deficiency of the lysosomal enzyme glucocerebrosidase. The enzyme deficiency or absence leads to multisystemic accumulation of glucosylceramide in macrophage lysosomes mainly of the spleen, liver, bone marrow and bone mineral, thus compromising the organ function [1].
Monoclonal gammopathy of undetermined significance (MGUS) is a premalignant plasma cell dyscrasia characterized by the presence of a stable serum monoclonal protein <3 g/dl and less than 10% clonal plasma cells in the bone marrow (BM) and by the absence of myelomarelated end-organ damage (CRAB - hypercalcemia, renal insufficiency, anemia, bone lesions) [2]. The rate of progression from MGUS to Multiple Myeloma (MM), Waldenstrom disease (WD), AL Amyloidosis and other lymphoproliferative disorders is approximately 1% of patients per year [3].
The correlation between GD and MGUS has already been described by several authors. The probable pathophysiology is based on the chronic B-cell antigen-driven stimulation by the augmented glycosilceramide or its deacylated products in the macrophages [4–6].
In the pediatric population MGUS is extremely uncommon and more frequently found in patients affected by other diseases, like GD, or in those who underwent solid organ transplantation [7,8].
Here, we describe a case of a 4-year old male patient affected by GD accompanied by IgG-k MGUS and successfully treated with biweekly imiglucerase in the absence of adverse events.
Case presentation
A 4-year old boy presented in March 2019 to our hospital with anemia and a palpable spleen (4 cm below the costal margin). The patient was born after full term pregnancy, had a steady growth with both height and weight charts within normal range and did not suffer from other medical conditions. The complete blood count showed microcytic anemia (hemoglobin 7.8 g/dL, MCV 67 fL) with normal white blood cell and platelet counts. Coagulation tests, liver and renal function tests, iron tests and hemoglobin electrophoresis were all normal. Furthermore, both the autoimmunity and infectious disease screening for HBV, HCV, HIV, Parvovirus B19, Mycoplasma Pneumonia and Leishmaina resulted negative. Serum protein electrophoresis revealed the presence of a small monoclonal protein of 0.1 g/dL, confirmed in immunofixation as IgG-k, with serum free light chain assay within normal range. No monoclonal protein was detected in the urine. An increased level of chitotriosidase (382 nmol/h/ml, r.v. 2-25) was observed. Enlarged spleen was confirmed with an abdominal ultrasound (spleen length: 13,5 cm). The patient was later discharged from the pediatric department in April 2019 due to his improved conditions and followed at our department for further diagnostic work-up in the suspect of Gaucher disease.
Figure 1:
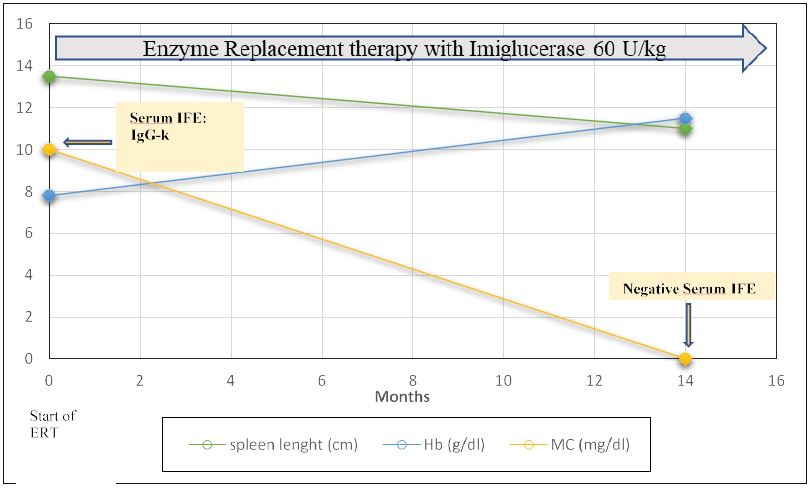
Figure 1:Treatment response in a pediatric patient with type 1 Gaucher Disease in course of Enzyme Replacement Treatment. ERT=Enzyme replacement treatment; Hb= hemoglobin; MC= monoclonal component; IFE: immunofixation.
Given the presence of anemia, splenomegaly and monoclonal protein together with increased chitotriosidase values, the patient was tested for GD in the setting of a prospective observational study on the prevalence of underrecognized type I Gaucher disease in a selected population with MGUS. The dried blood spot method (DBS) was used as screening and revealed low level of betaglucocerobrosidase enzyme, that was later confirmed by biochemical assay in peripheral blood leukocytes (1.1μmol/ L/h, reference values (r.v.). ≥ 4.1) and glucosylsphingosine (lyso-Gb1) quantification (389 ng/ml, rv ≤ 6.8) at another facility (Centogene). The genetic test revealed GBA1 mutation (double heterozygous N370S/L444P), thus confirming the diagnosis of Gaucher disease type I. Furthermore, he had low vitamin D level (9 μg/L, r.v. 30-100).
The patient was eligible for treatment due to its clinical presentation, including anemia, splenomegaly and MGUS. Therefore, in June 2019 he was eventually started on biweekly infusions with recombinant enzyme replacement therapy (ERT), imiglucerase (Cerezyme®, Sanofi-Genzyme), at a dosage of 60 U/kg in outpatient setting, together with vitamin D supplementation. The hemoglobin level improved in a matter of months, with complete recovery after six months of therapy. As for the IgG-kappa monoclonal protein, the immunofixation resulted negative after 1 year of imiglucerase biweekly infusions. There was also an initial reduction of spleen volume, evidenced in physical examination, with spleen palpable 2 cm below the costal margin in July 2020, and a follow-up abdominal ultrasound in December 2020 that confirmed spleen reduction (spleen length: 11 cm) (Figure 1). Vitamin D levels increased in course of supplemental treatment and the patient was evaluated for bone densitometry in December 2020 (Z score -1,2).
The patient is still in course of treatment with imiglucerase at the same dosage, without any record of adverse events.
Discussion
GD is a rare autosomal recessive inherited disorder caused by the deficiency of the lysosomal enzyme glucocerebrosidase, which results in the accumulation of its substrates in lysosomal macrophages (“Gaucher cells”) that are mostly found in the spleen, liver and bone marrow. Hepatosplenomegaly, anemia, thrombocytopenia, skeletal complications (pain, bone crises, avascular necrosis and fractures) and neurological symptoms represent the main clinical features. GD is traditionally classified into three forms depending on the age onset and the presence of neurological involvement. Type 1 GD (non-neuronopathic) can appear at any age and is characterized by the lack of involvement of central nervous system (CNS) [9]. It accounts for more than 90% of patients and the world-wide prevalence is 1 in 50.000-100.000 although in individuals of Ashkenazi heritage it is as high as ~1 every 850 people [10]. Because of its rarity and heterogenous clinical presentation, many patients experience important delay in the diagnosis, which results in severe complications that are today preventable or reversible by ERT or substrate reduction therapy (SRT), if started on time.
Polyclonal and monoclonal gammopathies are commonly found in patients with Gaucher disease with the prevalence ranging from 25 to 91% and from 0 to 35%, respectively [11]. The correlation between MGUS/MM and Gaucher disease has already been described in the literature [12-17]. Increased risk of haematological cancer and in particular of MM might be explained by both the direct oncogenic effect of the accumulated glycosphingolipids and the immunedysregulation in the bone marrow microenviroment [6]. It has been shown that monoclonal antibodies in GD-1 patients are frequently reactive against disease-related lysolipids, like lyso-glucosylceramide (LGL1) and lysophosphatidylcholine (LPC) [4]. This suggests that the chronic antigenic stimulation of the B lymphocytes by the accumulated glycosphingolipids can be responsible for the clonal evolution of the plasma cells and subsequent progression to MGUS and MM. The potential role of ERT and/or SRT on the onset and prognosis of MGUS and MM in GD patients still needs to be clarified. It has been reported that ERT results in an improvement in polyclonal gammopathies, but not in MGUS and its progression to MM [18]. On the other hand, SRT was able to prevent GD associated B-cell malignancy in mice [5] and the remission of MGUS in a GD-1 patient treated with ERT was recently reported [19]. However, further studies are needed in order to understand whether early treatment could influence the onset of malignancies.
Here we reported a case of a 4-year old male patient diagnosed with type I GD due to the presence of MGUS, together with anemia and splenomegaly, in the setting of a prospective observational study on the prevalence of underrecognized type I Gaucher disease in a selected population with MGUS.
In this clinical case, the presence of MGUS together with other more suggestive symptoms - splenomegaly and anemia – was of aid in order to perform GD screening. This led to an early detection of the disease and allowed the patient to start replacement treatment, thus preventing long-term complications. It should be said that in our case disappearance of the monoclonal protein was observed after one year of treatment, thus likely reducing the risk of hematological malignancies like multiple myeloma and other lymphoproliferative disorders. Probably, the prompt start of ERT contributed to the disappearance of the monoclonal clone.
Conclusion
Considering the good impact of an early recognition of the disease, hematologists should consider the possibility of concurrent GD in patients with MGUS, even in the absence of overt GD symptoms [20], especially given the availability and low-cost of current screening tests.

View pdf
Download pdf
Giuffrida G, Cappellini F, Carubbi M, Di Rocco G (2014) Management of bone disease in gaucher disease type 1: Clinical Practice Adv Ther 31: 1197-1212. [ Ref ]
Kyle RK, Durie BG, Rajkumar S, Landgren J. Blade G, et al. (2010) Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 24: 1121-1127. [ Ref ]
Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, et al. (2009) Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study, Blood113: 5412-5417. [ Ref ]
Nair S, Branagan AR, Liu J, Boddupalli CS, Mistry PK, et al. (2016) Clonal immunoglobulin against lysolipids in the origin of myeloma. N Engl J Med 374: 555-561. [ Ref ]
Pavlova EV, Archer J, Wang SZ, Dekker N, Aerts JM, et al. (2015) Inhibition of UDP-glucosylceramide synthase in mice prevents Gaucher disease-associated B-cell malignancy. J Pathol 235: 113-124. [ Ref ]
Costello R, O’Callaghan T, Sébahoun G (2006) Gaucher disease and multiple myeloma, Leuk. Lymphoma. 47: 1365-1368. [ Ref ]
Karafin MS, Humphrey RL, Detrick B (2014) Evaluation of monoclonal and oligoclonal gammopathies in a pediatric population in a major urban center, Am. J. Clin. Pathol. 141: 482-487. [ Ref ]
Gerritsen E, Vossen J, Van Tol M, Jol-Van Der Zijde C, et al. (1989) Monoclonal gammopathies in children, J. Clin. Immunol. 9 (1989) 296– 305. [ Ref ]
Andrade-Campos M, Alfonso P, Irun P, Armstrong J, Calvo C, et al. Diagnosis features of pediatric gaucher disease patients in the era of enzymatic therapy, a national-base study from the spanish registry of gaucher disease, Orphanet J Rare Dis 12. [ Ref ]
Grabowski GA (1997) Gaucher disease: Gene frequencies and genotype/ phenotype correlations, Genet. Test. 15-12. [ Ref ]
Arends M, Van Dussen L, Biegstraaten M, Hollak CE (2013) Malignancies and monoclonal gammopathy in gaucher disease; a systematic review of the literature J Haematol 161: 832-842. [ Ref ]
Fost M, Out TA, Wilde FA, Tjin EP, Pals ST (2008) Immunoglobulin and free light chain abnormalities in Gaucher disease type I: Data from an adult cohort of 63 patients and review of the literature, Ann. Hematol. 87 (2008) 439–449. [ Ref ]
Grosbois B, Rose C, Noël E, de R. Serratrice E, Dobbelaere D, et al. (2009) Gaucher disease and monoclonal gammopathy: A report of 17 cases and impact of therapy. Blood Cells Mol Dis 43: 138-139. [ Ref ]
Monge J, Chadburn A, Gergis U (2020) Synchronous multiple myeloma and Gaucher disease. Hematol Oncol Stem Cell Ther. 13 (2020) 42-45. [ Ref ]
Brady K, Corash L, Bhargava V(1997) Multiple myeloma arising from monoclonal gammopathy of undetermined significance in a patient with Gaucher’s disease. Arch Pathol Lab Med 121: 1108-1111. [ Ref ]
Harder H, Eucker J, Zang C, Possinger K, Muller-Hocker J, et al. (2000) Coincidence of Gaucher’s disease due to a 1226G/1448C mutation and of an immunoglobulin G lambda multiple myeloma with Bence-Jones proteinuria. Ann Hematol 79: 640-643. [ Ref ]
Pratt PW, Kochwa S, Estren S (1968) Immunoglobulin abnormalities in Gaucher’s disease. Report of 16 cases Blood 31: 633-640. [ Ref ]
Brautbar A, Elstein D, Pines G, Abrahamov A Zimran A (2004) Effect of enzyme replacement therapy on gammopathies in Gaucher disease. Blood Cells Mol Dis 32: 214-217. [ Ref ]
Camou F, Viallard JF (2012) Extended remission of B-cell lymphoma with monoclonal gammopathy in a patient with type 1 Gaucher disease treated with enzyme replacement therapy. Blood Cells Mol Dis 48: 51-2. [ Ref ]
Weinreb NJ, Mistry PK, Rosenbloom BE, Dhodapkar MV (2018) Lymphoplasmacytic malignancies, and gaucher disease: The significance of the clinical association Blood 131: 2500-2501. [ Ref ]
