Journal Name: Journal of Clinical Case Reports and Trials
Article Type: Case Report
Received date: 01 May, 2018
Accepted date: 22 August, 2018
Published date: 27 August, 2018
Citation: Abdelfadil AM (2018) Growth Hormone Therapy in a Case with Fanconi-Biekel Syndrome. J Clin Case Rep Trials. Vol: 1, Issu: 2 (08-13).
Copyright: © 2018 Abdelfadil AM. This is an openaccess article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Fanconi-Bickel syndrome is a rare and easily diagnosable disease, characterized by glycogen accumulation in the liver and kidneys and causing proximal tubular dysfunction and impair glucose and galactose utilization. I reported a 5 years old girl as a case of FBS presented with delayed walking, severe growth retardation with markedly short stature, doll-like facies, severe hypophosphatemic vitamin D resistant rickets, severe abdominal distension, hepatomegaly, renomegaly, and laboratory evidence of proximal tubular dysfunction. After the start of phosphate therapy, the patient can walk and improved in short time to normal gait, and her abdominal distension became partially improved. I started growth hormone therapy after addressing all the information regarding its cost, side effects, prognosis and possible little effectiveness of the hormone in this condition to the mother. Unexpectedly, I found good growth velocity ~ 8 cm/yr with the use of growth hormone in this case. Conclusion: growth hormone is effective in cases of FBS.
Keywords
Fanconi-Bckel syndrome, Rickets, Hepatomegaly, Growth hormone, Growth velocity.
Abstract
Fanconi-Bickel syndrome is a rare and easily diagnosable disease, characterized by glycogen accumulation in the liver and kidneys and causing proximal tubular dysfunction and impair glucose and galactose utilization. I reported a 5 years old girl as a case of FBS presented with delayed walking, severe growth retardation with markedly short stature, doll-like facies, severe hypophosphatemic vitamin D resistant rickets, severe abdominal distension, hepatomegaly, renomegaly, and laboratory evidence of proximal tubular dysfunction. After the start of phosphate therapy, the patient can walk and improved in short time to normal gait, and her abdominal distension became partially improved. I started growth hormone therapy after addressing all the information regarding its cost, side effects, prognosis and possible little effectiveness of the hormone in this condition to the mother. Unexpectedly, I found good growth velocity ~ 8 cm/yr with the use of growth hormone in this case. Conclusion: growth hormone is effective in cases of FBS.
Keywords
Fanconi-Bckel syndrome, Rickets, Hepatomegaly, Growth hormone, Growth velocity.
Abbreviations
BCM: Below Costal Margin, FBS, Fanconi-Bickel syndrome.
Introduction
Fanconi-Bickel syndrome is a rare but a well-defined clinical entity inherited in an autosomal recessive mode [1] and characterized by hepatorenal glycogen accumulation, proximal renal tubular dysfunction, and impaired utilization of glucose and galactose [2]. Its name came for Guido Fanconi and Horst Bickel, who first described it in 1949 [3,4]. Use of the term glycogenosis type XI introduced by Hug is to be discouraged because glycogen accumulation is not due to the proposed functional defect of phosphoglucomutase, an essential enzyme in the common degradative pathways of both glycogen and galactose, but is secondary to nonfunctional glucose transport [5].
Inheritance
Fanconi-Bickel syndrome (FBS) is an autosomal recessive genetic disease due to single-gene disorder caused by a mutation in the SLC2A2 gene (OMIM 227810) gene mapped on chromosome 3q26.1-26.3 [6]. This genetic defect results in defective formation of a protein called glucose-transporter protein 2 (GLUT2) [7-9]. This protein is responsible for transporting glucose in and out through different cells in the body. When GLUT2 is not working properly because of a mutation in SLC2A2, the body cannot transport glucose. Therefore, glucose builds up and stored in the liver and kidneys as glycogen. The buildup of glycogen in these body parts causes the symptoms of Fanconi Bickel syndrome [7-10]. More than 30 different mutations have been identified, and most of the reported mutations are private and confined to a single family [11].
Case-Report
Rodina is 5years old girl, born in Bani swif city in Egypt at 6 of July, 2011 with Lower Section Caesarean Section due to the prolonged 2nd stage of labor, she was full term at delivery, and her parents are consignees to 2nd degree consanguinity, divorced parents since the mother was pregnant. Mother came with the complaint of delayed walking, short stature & abdominal distension since early infancy period. Parents observed abdominal distension since the 1st month of life that was increasing in degree till reach the present status, mother asked for medical advice and prescribed different medications that were not effective. Delayed walking till the age of 6 years with no H/O impaired motor development as good head support at 2-3 month, sitting with support at 4-5 month while sitting without support at 6-7 month, at 9-10 month crawling while sitting, normal crawling at 14 mo. At the age of 11 months, she suffered fracture of both femurs during a trial to walk followed by malunion and at the age of 2 years, she suffered from the 2nd fracture of the right lower tibia. She had developed deformity of both legs in the form of anterior convexity in the middle of both femurs and at the lower third of the Rt tibia, as seen in the pictures and X-Rays of lower limbs, teething with lower central incisors at 10 month and upper central incisors at 14 months. She was investigated and started one alpha (active form of vitamin D) and L-Carnitine with different calcium preparations without improvement since the age of 2 years till the time of the diagnosis.
Mental development is normal with a history of social smile at age of 1 month, babbling from the 2nd month while the 1st word at the age of 4th month; telegraphic sentence at age of 1-1.5 year; can tell stories at age 3rd year.
Social development also is normal, she can hold spoon & feed herself at the age of 1 year, control urination at 18 month and control stools at 2 years, playing with toys since the age of 7 months and enjoy playing with other friends at the age of 7 months. She was investigated and started one alpha (active form of vitamin D) and L-Carnitine with different calcium preparations without improvement since the age of 2 years till the time of the diagnosis.
Past history is irrelevant with free prenatal history and the usual follow up with an obstetrician; who reported normal intrauterine growth, full-term delivered LSCS, didn’t need resuscitation at birth and didn’t need admission to the neonatal intensive care unit.
Vaccination: Vaccinated up to date; all compulsory vaccination (oral Polio, DPT, Hepatitis B, Measles, MMR) according to the Egyptian schedule of immunizations.
Family history is irrelevant with no history of similar conditions.
Examination
Generally, looks well & good, with a doll-like faces, smiling, but poorly thriving, pink in room air, hydrated well, playful mood, and talkative (Figure 1).
Figure 1
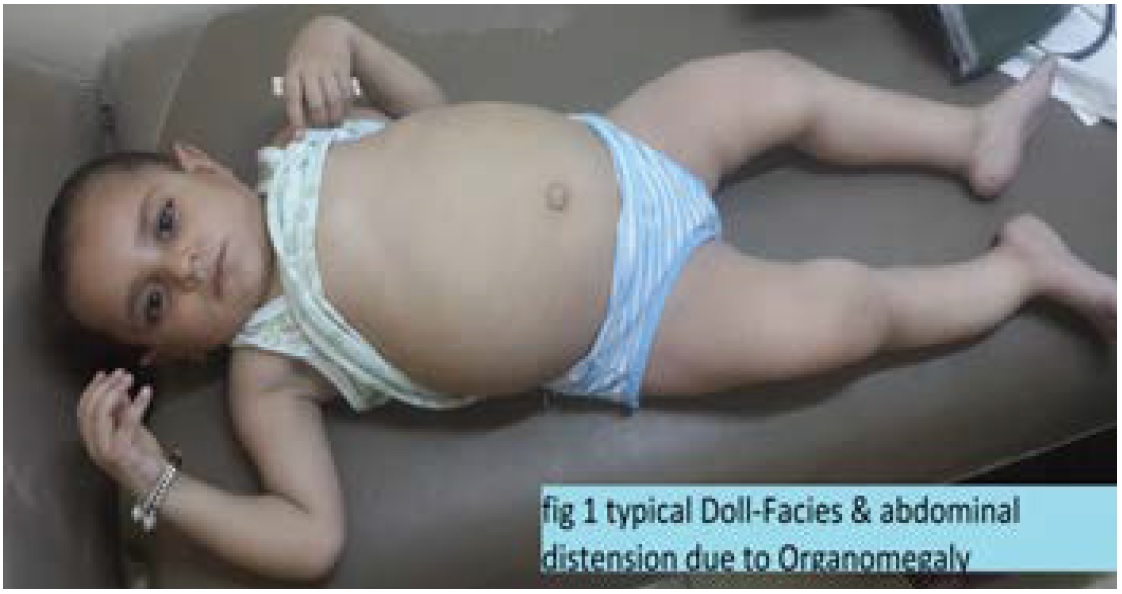
Figure 1: Typical Doll-like Facies with abdominal Distension due to Organomegaly.
Vital signs: Heart rate 80/min, respiratory rate 20/min, blood pressure 100/60 while recumbent.
Anthropometric measurements at age of 4 yrs. 10mo; her weight was on -2.09 SD, height was on -5.9 SD while recumbent; BMI 19.8, HC 47 cm (Figures 2 and 3). Upper segment 49cm, Lower segment 33 cm with anterior curvature deformity of both femurs, U/L segment ratio 1.4. Skin is free with no skin rash.
Figure 2
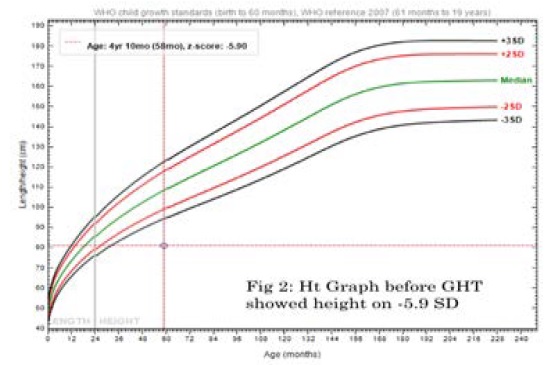
Figure 2: Height Graph before Growth Hormone Therapy with height on -5.9 SD.
Figure 3
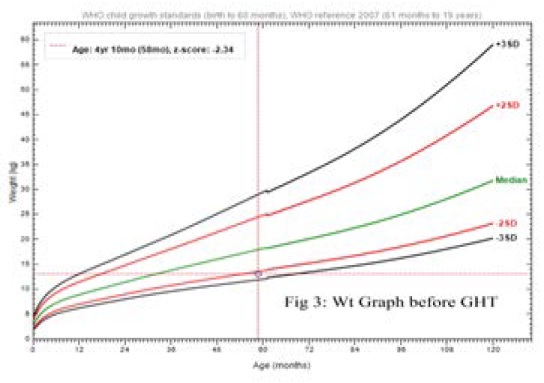
Figure 3: Weight graph with weight on -2.09 SD.
Rackitic signs as visible rosary beads & Harrison sulcus, no Marfan sign, teeth are well and complete dentition with no dental caries, bilateral genu varum. Mild muscle wasting of 4 limbs.
Cardiovascular system examination is free with normal & equal peripheral pulsations, no cardiomegaly, normal S1&S2 and no murmur.
Chest examination: no respiratory distress, normal bilateral equal air entry, no adventitious sounds
Abdominal examination: marked abdominal distension with distension of flanks & shifting of the umbilicus downward with no visible veins; by palpation: tense abdomen, but after phosphate therapy became lax, liver is hardly felt around 18 cm BCM, firm in consistency, not tender, smooth surface, rounded border; no palpable spleen, no shifting dullness, and normal female genitalia with normal back and spine examination.
Neurologic examination: mild generalized muscle wasting with normal muscle tone in the 4 limbs and trunk, and normal deep tendon reflexes. Normal eye examination with normal fundus.
Cerebellar function is good.
Radiologic investigations
Abdominal ultrasound: hepatomegaly with liver span around 18 cm, no ascitis, normal spleen with average size and no focal lesions; both kidneys are of average size with no stones, cysts and no back pressure.
Plain X Rays of the left hand and wrist for calculating bone age with Greulich and Pyle method: bone age is equivalent to 2 6/12 years, with active rachitic changes (Figure 4).
Figure 4
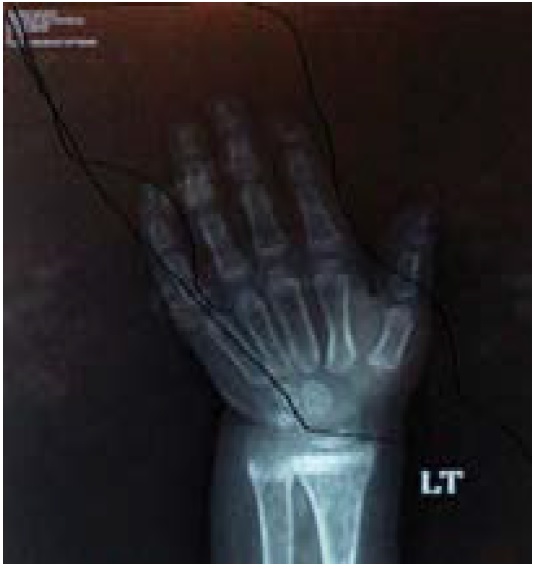
Figure 4: X-rays of left hand and wrist for bone age: showed rackitic changes and delayed bone age of 2 – 21/2 years.
X Rays of right ankle (A.P and Lateral view at 22/6/2014): Malunited old fracture of the lower tibia with anterior curvature within the markedly osteopenic texture with foraging and widening of metaphysical lining denoting rachitic changes (Figure 5).
Figure 5
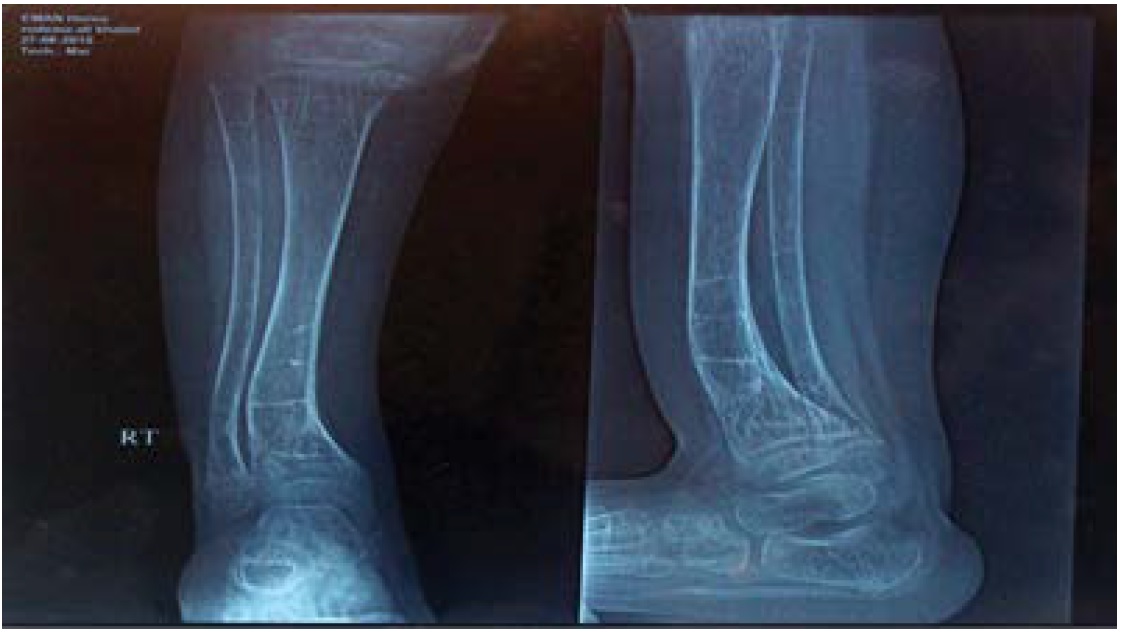
Figure 5: X-rays of right lower leg and ankle joint showed old fracture lines, osteoporosis, rackitic changes and deformity of the lower tibia with anterior curvature.
Laboratory investigations
CBC: HB 13.5 g/dl, HCT 40%, MCV 91.9 fl, MCH 30, MCHC 33.5%, WBCs 6.9 Χ 109/l, Neutrophil count 1.8 Χ109/l (26.4%), Lymph 4.7 Χ109/l (67.4%), Monocytes 0.4 Χ109/l (6.2 %), Platelet count 347 Χ109/l, MPV 6.9 fl, PDW 8.6 fl,
Liver function tests: SGPT 34, SGOT 65, Albumin, Cholesterol 336, Direct Bilirubin, PT 11.5, PC 100%. Lipid profile was normal.
Urea 39 mg/dl (15-40 mg/dl), Creatinine 0.2 mg/dl (0.6-1.2 mg/dl). Serum potassium 2.6 mg/dl (3.5-5 mg/dl), serum Sodium 135 mg/dl (135-145 mg/dl).
Uric acid level 2.3 mg/dl (2- 2.7 mg/dl)
Normal Anion gap metabolic acidosis with pH 7.27, bicarbonate 12, and a base deficit-6.6.
Blood sugar: Fasting hypoglycemia 40 mg/dl, 2 hrs postprandial: 120 mg/dl.
Vitamin D (1, 25 Dihydroxy-cholecalciferol): 21.3 pg/ml (18-78).
Ant mitochondrial antibodies: negative (by IF immunofluorescence technique).
Anti-Smooth muscle antibodies: positive 1/20h (by indirect immunofluorescence test).
Anti-Liver-Kidney Microsomal antibodies: negative (the starting dilution of LKM is 1/20).
Total IgA: 66.9 mg/dl.
Anti-Human Tissue Transglutaminase: negative 4.3 EU/ ml (by ELISA) (Table 1).
Table 1: Serum Calcium, Phosphorus, Alkaline phosphatase and Active Vitamin D Level in F/U visits.
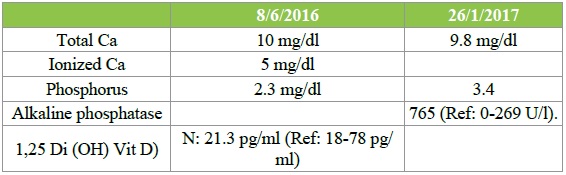
Ca: Total: 10 mg/dl repeated was the same, Ionized 5 mg/dl (1.25 mmol/l).
Phosphorus is low 2.3 mg/dl (3.4-6.2mg/dl) when repeated was same. When repeated under treatment with phosphorus therapy it was 3.4 mg/dl.
Alkaline phosphatase: H 765 U/l (0- 269).
Urine analysis: pH 4.6, aminoaciduria, glucosuria (+3), and phosphaturia.
Liver biopsy: consistent with glycogen storage disease with diffuse ballooning of hepatocytes with abundant clear or granular cytoplasm. Nuclei are rounded and small, there is infiltration of portal tracts with mature lymphocytes. No evidence of piecemeal necrosis, no evidence of portal bridging or cirrhosis, and no evidence of dysplasia or malignancy.
Karyotype: normal female karyotype, 46 XX.
Now, the patient walks well and can run with stable gait and muscles of lower limbs acquire good girths.
The patient has been treated with the Joulie’s solution that is consisted of Dibasic Na+ Phosphate 136 gm+ Phosphoric Acid 58.8 gm dissolved in 1 liter of tap water. Each 1 ml=30 mg phosphorus.
The dose will be 30-50 mg/kg/day.
The dose is calculated and constituted from both derivatives and dissolved in tap water that will be equivalent to the contents. This solution will be stable only for 2 weeks.
Growth Hormone Therapy (GHT): Investigations done before the start of GHT: karyotype, x-rays of left hand and wrist for assessment of bone age by Greulich and Pyle method, IGF1; done by radioimmune assay and was 61 nmol/L that was normal for age and sex.
The Random Growth hormone level was 750 pmol/L (N), but Growth hormone stimulation test is not done because of expected severe hypoglycemia and its consequence.
GHT has been started since Jan 15, 2017 by a dose of 25ug/kg/d is given till nowadays. The height had improved from 81cm (-5.90 SD) to 88.5 cm (-5.23 SD) at July 15, 2017, with growth velocity of 8 cm/yr.
On the same dose of GH, the height increased at the age of 76 mo to 90.2 cm (-5.13 SD) at Nov 4, 2017, and also, the height increased to 94 cm (-4.9 SD) at June 6, 2018 at the age of 83 mos of life.
growth velocity is around 10 cm/year (Figure 6).
Figure 6
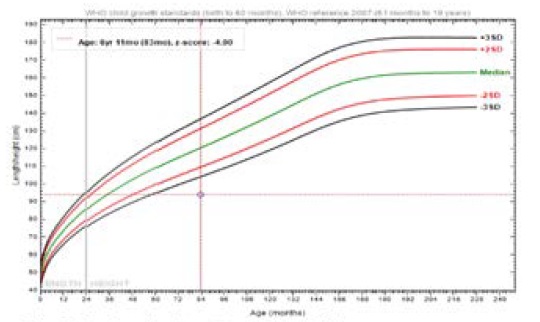
Figure 6: Height Graph after 14 months of GHT.
Prophylactic dose of vitamin D: 600 iu daily as the level of 1.25 di-hydroxy vitamin D is normal.
Potasium level was maintained normal; no K supplementation given.
The mother is recommended to give frequent carbohydrate feeds and corn starch at bedtime.
Discussion
Although FBS is a rare syndrome, but its clinical presentation and laboratory findings make it as a clear clinical entity. About 115 cases have been reported in the literature, from different parts of the world; including the Mediterranean belt, Turkey, Japan, Europe, Arab countries of the Near East and North Africa and North America [3,4,6].
The age of presentation of FBS is between the 3rd to 10th month with failure to thrive, severe hypophosphatemic rickets, “doll-like” face, and severe abdominal distension due to massive hepatomegaly [3,4].
Our patient presented later because of previous wrong diagnoses as vitamin D deficiency rickets with dyspepsia at many times and given frequent courses of active vitamin D and vitamin D3 plus symptomatic treatment of suspected gaseous abdominal distention as she was distended to the degree that you cannot feel organomegaly by abdominal examination.
Laboratory findings of FBS that of proximal tubular nephropathy which presented by glucosuria, generalized aminoaciduria, bicarbonate wasting, phosphaturia, and hypophosphatemia plus fasting hypoglycemia, hyperglycemia and hypergalactosemia in the post absorptive state indicating an impaired utilization of these two monosaccharides, and hyperlipidemia may be present [3,4].
After the clinical suspicion of my case that is confirmed by other lab results as above, abdominal ultrasound confirmed the presence of both hepato and renomegaly.
The Kidneys become noticeably enlarged by two years of age [12].
Clinical findings in the patient of failure to thrive, short stature, severe hypophosphatemic rickets with improvement of Jouli’s solution, and severe abdominal distension due to hepatolmegaly that is due to glycogen storage disease proved by Liver biopsy, addition to radiological and laboratory findings confirms the case is Fanconi-Bickel Syndrome
There is no specific therapy available for this condition. Free access to water to avoid dehydration from polyuria and symptomatic replacement of phosphate, electrolytes, sodium bicarbonate and vitamin D, and restriction of galactose, is recommended. FBS patients absorb simple sugars like glucose and galactose poorly in their diet. However, they can absorb complex carbohydrates like uncooked corn starch [2,13]; so fasting should be avoided and the patient should be given mild frequent feeds and uncooked starch at bedtime to prevent hypoglycemia; this also helps in regression of hepatomegaly [3,4].
FBS patients typically enter puberty later than expected with weakened bones resulting from osteopenia or osteoporosis [14], they will become shorter than other people as adults, and they may have bowed legs [8].
As regards to the differential diagnosis, von Gierke’s disease is considered to be nearest one. These patients usually have doll-like faces, biochemical abnormalities like hypoglycemia, hyperuricemia and hyperlipidemia besides defective glucose-6-phosphatase enzyme. Later in life they may manifest renal abnormalities like nephrocalcinosis, distal renal tubular acidosis and, rarely, Fanconi syndrome [15-17].
Nobody in the literature used growth hormone therapy in case of FBS. The use of growth hormone was started after giving instructions to the mother that growth hormone therapy may not be effective due to the expected easy loss of the hormone through the kidneys due to Fanconi syndrome. In spite of that, growth velocity unexpectedly was improved to reach 8 cm/yr and improvement of the height from-5.7 to-5.23 within 8 months of therapy.
Conclusion
FBS can be easily diagnosed due to its distinct clinical and laboratory findings and get benefit from growth hormone therapy.
Declaration
Acknowledgments
To all medical personnel including doctors and nurses who are working in Bani Suif Insurance Hospital, both internal department and OPD, and to Al-Borg Clinicopathological lab.
Funding
Personally funded, no financial support from any institutions.
Competing interests
Author has declared that there are no competing interests.
Consent for publication
Written consent from the mother is available.
Ethics approval and consent to participate
The study was carried out according to the principles of declarations of Helsinki, and its appendices [17] and was approved the hospital ethical review board in El Minia university hospital (code 75a, March, 2015). Written informed consents from patients’ caregivers were obtained for the use of their study-related information and for participation in the ongoing research.

View pdf
Download pdf
There is no references
