Journal Name: Journal of Multidisciplinary Research and Reviews
Article Type: Review
Received date: 12 November, 2018
Accepted date: 22 November, 2018
Published date: 29 November, 2018
Citation: Sedzro DM, Bellah SMF, Akbar H, Billah SMS (2018) Structure, Function, Application and Modification Strategy of ? –Galactosidase. J Multidis Res Rev. Vol: 1, Issu: 1 (10-16).
Copyright: © 2018 Sedzro DM. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Also called lactase, β-galactosidase (beta-gal or β-gal), is a glycoside hydrolase enzyme that catalyzes the hydrolysis of β-galactosides into monosaccharides through the breaking of a glycosidic bond. Carbohydrates containing galactose where the glycosidic bond lies above the galactose molecule are an example of β-galactosides. Ganglioside GM1, lactosylceramides, lactose, and various glycoproteins are different substrates of β-galactosidases. In both the history and the practice of molecular biology, β-Galactosidase (Escherichia coli) has a special place. In Jacob and Monod’s development of the operon model for the regulation of gene expression, it played a fundamental role. By producing an easily recognizable blue reaction product, it has the ability to signal its presence which has made it a workhorse in cloning and other such molecular biology procedures. The purpose of this review is to provide an overview of the enzyme function, enzymatic mechanism (chemistry and structure), application in biotechnology industry, advantages and disadvantages, and modification strategy of β-galactosidase.
Keywords
Lactase, β-galactosides, Escherichia coli, Ganglioside, Lactosylceramides.
Abstract
Also called lactase, β-galactosidase (beta-gal or β-gal), is a glycoside hydrolase enzyme that catalyzes the hydrolysis of β-galactosides into monosaccharides through the breaking of a glycosidic bond. Carbohydrates containing galactose where the glycosidic bond lies above the galactose molecule are an example of β-galactosides. Ganglioside GM1, lactosylceramides, lactose, and various glycoproteins are different substrates of β-galactosidases. In both the history and the practice of molecular biology, β-Galactosidase (Escherichia coli) has a special place. In Jacob and Monod’s development of the operon model for the regulation of gene expression, it played a fundamental role. By producing an easily recognizable blue reaction product, it has the ability to signal its presence which has made it a workhorse in cloning and other such molecular biology procedures. The purpose of this review is to provide an overview of the enzyme function, enzymatic mechanism (chemistry and structure), application in biotechnology industry, advantages and disadvantages, and modification strategy of β-galactosidase.
Keywords
Lactase, β-galactosides, Escherichia coli, Ganglioside, Lactosylceramides.
Enzyme Function
β-galactosidase has three enzymatic activities as shown in Figure 1. Foremost, it can cleave the disaccharide lactose to form glucose and galactose, which can then enter glycolysis. Secondly, the enzyme can catalyze the trans-galactosylation of lactose to allolactose, and, finally, the allolactose can be cleaved to the monosaccharides. It is allolactose that binds to lacZ repressor and creates the positive feedback loop that regulates the amount of β-galactosidase in the cell [1].
Figure 1
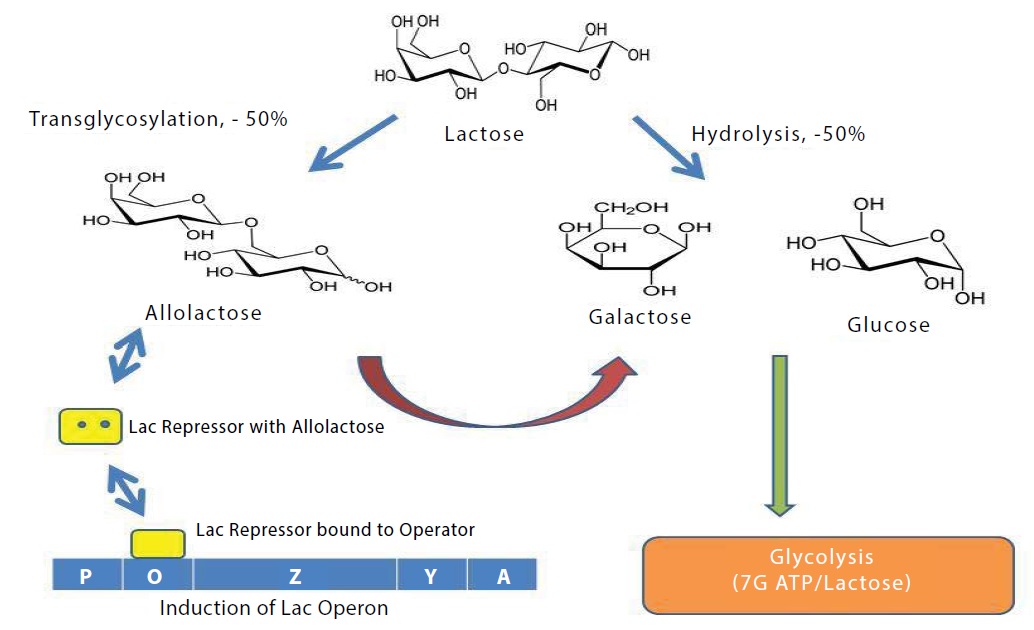
Figure 1: Diagram summarizing the functions of β-galactosidase in the cell. The enzyme can hydrolyze lactose to galactose plus glucose, it can transgalactosylate to form allolactose, and it can hydrolyze allolactose. The synthesis of allolactose which binds to the lac repressor and reduces its affinity for the lac operon is as a result of the presence of lactose. This in turn allows the synthesis of β-galactosidase, the product of the lacZ gene.
X-gal (5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside), a soluble colorless compound consisting of galactose linked to a substituted indole reacts with β-galactosidase and the latter is best recognized for this interaction. There is high specificity for the galactose part of β-galactose substrates but low specificity for the remainder. Thus, an insoluble intensely blue product is produced when it hydrolyzes X-gal, releasing the substituted indole that spontaneously dimerizes. On growth medium containing X-gal, colonies of E. coli that have an active β-galactosidase become blue because of this reaction.
In single crystals of the enzyme, the X-gal reaction can readily be performed. As proteins in general, β-Galactosidase form crystals that include about 50% protein and 50% solvent by volume. Extending throughout the crystal, the solvent-filled channels are much larger than the substrate and allow substrate to freely diffuse throughout the crystal. Wyckoff et al. in early experiments on the nature of protein crystals, used a flow cell to investigate the diffusion of ligands into a 0.4 mm crystal of ribonuclease S [2]. The half-time for re-equilibration within the crystal was 90 s when the concentration of ammonium sulfate surrounding the crystal was rapidly changed [2].
Matthews [3] obtained very similar results when he used crystal density measurements to monitor the diffusion of ammonium sulfate solutions into crystals of γ-chymotrypsin. It can be estimated based on these experiments that a molecule the size of X-gal will diffuse through a 0.4 mm × 0.4 mm × 0.4 mm crystal of β-galactosidase in several minutes.
The blue color of a crystal of β-galactosidase exposed to X-gal in confirms that the enzyme in the crystal is catalytically competent. It also tends to suggest, but does not prove, that catalysis proceeds via relatively modest changes in the conformation of the enzyme, that is, there is no suggestion of major structural changes which might destroy the crystals.
A novel procedure to measure the activity of single β-galactosidase molecules has been developed by Craig and co-workers [4]. The procedure depends on the conversion of the weakly fluorescent substrate resorufin β-d-galactopyranoside to the highly fluorescent product resorufin. Using a typical incubation of 15 min, the amount of product molecules can be measured with an estimated error of about 15% since individual β-galactosidase molecules produce several thousand product molecules per minute.
Using a purpose-designed capillary electrophoresis instrument the single-molecule activity measurements are performed. The instrument has a number of advantages. For instance, a single protein molecule can be allowed to react with substrate for a desired period, and then moved away from the accumulated product into a new location, permitting a repeated measurement with the same protein molecule. Another advantage is that activity measurements for several protein molecules can be performed in a single experiment. Molecules of β-galactosidase displayed a range of activity of 20-fold or greater both before crystallization, and from dissolved crystals. Molecules from a crystal had an overall activity distribution of 31,600 ± 1100 reactions per minute while the pre-crystallized protein had an overall activity distribution of 38,500 reactions per minute [5].
On one hand, it could be argued that the range of catalytic activities reflects individual β-galactosidase molecules oxidation or other such chemical modification. The crystallized proteins have slightly lower activity than those measured before crystallization and this could be due to additional chemical modification during crystal growth. On the other hand, the observed distribution of activities in the above scenario is not easily rationalized [5]. It might be anticipated that a relatively large population of “undamaged” β-galactosidase molecules with identical activities would be present and the “damaged” molecules would then have a range of lower activities. This was not observed at all.
Shoemaker et al. [5] indicated that the bulk of the molecules (ca. 80%) have activities between 11,000 reactions/min and 50,000 reactions/min while a smaller number (ca. 20%) have activities that extend up to at least 100, 000 reactions/min. The origin of these “superactive” molecules is not obvious and Shoemaker et al. [5] assert that one possible explanation is the presence of higher oligomeric forms of the enzyme. In this assessment, a β-galactosidase octamer, for example, with eight active sites, would still be counted as a single molecule. On the other hand, if some molecules were tetramers and others octamers, these would be expected to give two distinct activity values.
Specificity
β-galactosidase catalyzes reactions with β-dgalactopyranosides with an oxygen glycosidic bond [6]. It also reacts, but with much reduced catalytic efficiency, with nitrogen, and sulfur and fluorine which are substrates having other glycosidic linkages [7].The enzyme is very specific for d-galactose [7] and the 2, 3, and 4 positions are especially important. For the enzyme to catalyze the reaction, the hydroxyls at those positions must each be present and in the correct orientation. The fraction of binding energy released as a result of interactions at the 2 position was much larger than the fraction contributed from interactions with the other galactosyl hydroxyls as demonstrated through studies [8] with fluorinated and deoxy glycosides.
Only d-galactopyranose, l-arabinopyranose, d-fucopyranose, and d-galactal reacted in the reverse direction when d-glucose was the other reactant. This was shown through reversion (reverse) β-galactosidase reactions [9] done at very high concentrations of sugars having orientation changes and/or the absence of individual hydroxyls at different positions. The enzyme also hydrolyzes p-nitrophenyl-α-larabinopyranoside and p-nitrophenyl-β-d-fucopyranoside (which do not have O6 hydroxyls), but these substrates bind poorly and react slowly [10].
Therefore, sugars with modifications at the C6 hydroxyl position of d-galactose are still substrates, albeit poor ones but sugars with changes elsewhere (except d-galactal) are unreactive. When Glu537 reacts with the C1 with a simultaneous proton addition at the C2 position, d-galactal forms a relatively stable covalent intermediate and that is the reason why it reacts [11]. This results in the formation of covalently attached 2-deoxy-galactose that is released on hydrolysis. In the reversion reaction [9] at high concentrations of d-galactal and glucose, the covalent entity reacts with glucose.
Enzymatic Mechanism (Chemistry and Structure)
β-Galactosidase is a tetramer of four identical polypeptide chains, each of 1023 amino acids [12]. In a monoclinic crystal form with four tetramers in the asymmetric unit (Figure 2), the crystal structure was initially determined [13]. Subsequently the structure was refined to an orthorhombic crystal with a single tetramer in the asymmetric unit [14]. Used for subsequent structural and functional studies, the latter form is technically superior and has been. Within each monomer the 1023 amino acids form five well-defined structural domains [13,14]. A so-called triose phosphate isomerase (TIM) or α8β8 barrel is the third (central) domain with the active site forming a deep pit at the C-terminal end of this barrel (residues 334–627). As noted below, critical elements of the active site are also contributed by amino acids from elsewhere in the same polypeptide chain as well as from other chains within the tetramer.
Figure 2
Figure 2: Beta-galactosidase from Penicillum sp (Adapated from Wikipedia).
Researchers have suggested that β-galactosidase arose from a much simpler, single-domain TIM barrel enzyme that had an extended active-site cleft and could have cleaved extended oligosaccharides [15]. The size of the active-site cleft to a pocket commensurate with binding disaccharide substrates could have been reduced by the subsequent incorporation of additional domains. Furthermore, the production of allolactose which is an inducer might have been promoted through some of these additional elements.
α-Complementation and Metal Requirements
It was found, in early studies of β-galactosidase, that deletion of certain residues near the amino-terminus such as 23–31 or 11–4110, [16] caused the tetrameric enzyme to dissociate into inactive dimers.
Additionally, it was possible to reconstitute the active tetrameric form of the enzyme by using peptides that included some or all the “missing” residues (e.g., 3–41 or 3–92) [17]. This phenomenon of “α-complementation” is the basis for the common blue/white screening (with X-gal) used in cloning. It can now be rationalized in terms of the three-dimensional structure.
Residues from about 13 to 20 in adjacent subunits contact each other as can be seen in Figure 2 [18]. A study of α-donors with substitutions showed that Glu17 is important for α-complementation. Occurring at the top of the figure is an equivalent interaction between the other two subunits. The tetramer dissociates into dimers (or “α-acceptors”) due to the removal of these residues which weakens the vertical activating interface. At the same time, the residues that form the horizontal long interface are unchanged and allow the protein to remain as dimers. The dimers are somewhat unstable and tend to dissociate to monomers unless thiols and sufficient Na+ and Mg2+ are present. These additives stabilize the dimeric structure [19].
When the complementation peptide (“α-donor”) is supplied to the α-acceptor, it binds at the site vacated by the removed N-terminal residues. This will further stabilize the binding of the α-donor and help restore the tetrameric structure. As long as the N-terminal ∼41 amino acid residues are present, the length of the α-complementing peptide is not important. Even denatured whole wild type enzyme brings about complementation [20].
There are major consequences for the activity of the enzyme in the above transition between the tetrameric and dimeric states. The active site includes critical catalytic residues of other domains although it is formed primarily by the TIM barrel of Domain. In particular, a loop from Domain 2 of Monomer A extends across the activating interface to contribute to the active site of Monomer D. In total, there are four such interactions across the activating interface (A to D, D to A, B to C, and C to B) that form the four equivalent active sites per tetramer. Contrariwise, all four active sites are disrupted by the dissociation of the tetramer into the dimer and completely abolish the activity of the enzyme.
To be fully active, β-Galactosidase requires Na+ or K+ [21] and Mg2+, [22]. Dependence on the monovalent and divalent cations is not absolute as there is some residual activity in their absence even though the cations are important for both binding and reactivity. As shown in, the ligands coordinating with Na+ or K+ are the carboxyl of Asp201, the peptide oxygen of Phe601 and the side-chain oxygen of Asn604, and between 1 and 3 waters. Moreover, of importance may also be the interaction between Tyr100 with Asp201. Interestingly, the π electron cloud of the benzyl group of Phe601 also becomes a “ligand” of the monovalent cation during the reaction. In addition, whenever substrate, transition states, or the covalent intermediate are at the active site, the O6 hydroxyl of galactose replaces one of the waters.
Seemingly unique to β-galactosidases are interactions such as this, between the monovalent ion and a hydroxyl. The functions of monovalent cations at the active sites of all other enzymes that are known to have monovalent metal requirements[23]either help neutralize the negative charge of substrate phosphate groups, often in conjunction with a divalent cation, or they have structural importance, but in this case, Na+ interacts with a hydroxyl group.
Initial Substrate Binding
Not totally buried, substrate initially binds in the active site in the shallow mode [24]. The dissociation constant, Ks, is associated with this interaction. The active site loop and Phe601 are open both before and after substrate binding but the open form of the loop and of Phe601 are defined a little better than the closed form by electron density when substrate is bound [25].This indicates that the loop is not fully open until substrate is bound.
With the galactosidic oxygen more or less centered over the indole8 [26] the substrate in the shallow mode takes up a position roughly parallel to Trp999. in the van der Waals range are the distances between Trp999 and most of the galactosyl and glucosyl carbons [27]. In these cases, facing towards the indole are the sides of the sugars having hydrogens rather than hydroxyls. These hydrogens are thought to have partial positive charges induced by electron pull by the hydroxyl groups on the opposite side. These partially charged hydrogens interact with the π electron cloud of the Trp999 indole. It is now seen that hydrophobic aglycones of synthetic substrates interact with Trp999 and were shown some time ago to bind strongly [24,28].
There are specific bonds to each of the hydroxyls of the galactosyl component of the substrate in the shallow site, in addition to the interaction with Trp999. Glu461 forms an H-bond (∼2.6Å) with the C2 hydroxyl while Glu537 is close enough (∼3.1 Å) to form an H-bond with the C3 hydroxyl. Asn460 forms an indirect interaction with the C3 hydroxyl (via water). [29]The C4 hydroxyl of the galactosyl portion of the substrate interacts with Asp201 and with a water molecule ligated by the active site Mg2+. There is a hydrophobic interaction between the C6 and the benzyl group of Phe601 and Asn604 interacts with the C6 hydroxyl. The C6 hydroxyl also seems to have strong interactions with both Na+, [24] and His540 [30].
Although there is an intralactose 2.9Å hydrogen bond between the C3 glucosyl hydroxyl and the galactosyl ring oxygen, except for contacts with Trp999, there are no specific bonds with the glucose portion of lactose. The o-nitro group of oNPG interacts with Trp999 but also forms an interaction with His418 [24]. There is, however, no bond between the p-nitro group of pNPG and His418. Because some of the intrinsic binding energy cannot be accessed, despite the extra bond, oNPG binds less well than pNPG. This interaction has significant effects on the galactosylation rate and the oNPG reaction rate is considerably different from the pNPG rate [31].
Application in Biotechnology Industry
Having natural built-in stability to temperature and other inactivation agents, β galactosidases from thermophile microorganisms could be useful both in solution and immobilized form in food industries, allowing for a simultaneous soft thermal treatment and the low hydrolysis of lactose [32,33]. Thermophilic β galactosidases enzymes have been used in the industrial processing of dairy products along with heat treatment to sterilize the product and represent a very useful alternative to the mesophilic enzymes. The lowering of microbial contamination is one of a number of generally recognized advantages in the industry with reference to the enzymatic treatment of these kinds of materials at high temperatures [34].
Cold-active β galactosidase, which hydrolyzes lactose to its basic components, is one of the important food-industrial enzymes. Psychrophilic yeasts, which grow on lactose as a sole carbon source at low temperature and under acidic conditions, were isolated from cold active β galactosidase. The Gram-positive Antarctic bacterium Arthrobacter sp. C2-2 contains two, possibly three cold-active isoenzymes of β galactosidase. The C2-2-1 isoenzyme was cloned, purified and characterized. Tis β galactosidase was classified as being a member of the family 2 of glucosidases [35].
An ultra-sonication method is widely used for laboratory scale work in biotechnological processes, and it does not require sophisticated equipment or extensive technical training. Ultrasound irradiation can change the structure and function of biological molecules. Therefore, the level of intensities of ultrasound plays the major part in the activity or inactivity of many enzymes after ultra-sonication.
Release of higher β galactosidase activity from lactic acid bacterial cells to the culture medium was as a result of sonication. However, only when β galactosidase was electively released was lactose hydrolysis enhanced. Much higher than those in conventional fermentation, the degree of lactose hydrolysis obtained was nearly 75% as compared to the usual degree below 40%. A suitable sonication method could simultaneously obtained a high viable cell count and a high degree of lactose hydrolysis as the findings of this work exhibited [36].
It has been found to be an interesting alternative for the lactose hydrolysis to use whole cells as a source of β galactosidase but a major drawback in the use of whole cells is the poor permeability of the cell membrane to lactose [37]. Permeabilization technology can overcome this problem and be helpful in the development of a low-cost technology for the hydrolysis of lactose. Thus, for the production of lactose-hydrolyzed milk using yeast cells, permeabilization technology was applied. Under optimized conditions, the ethanol permeabilized yeast cells gave 89% hydrolysis of milk lactose [38].
After hydrolysis, dairy whey which is a contaminant product with a high organic chemistry demand, may be used as cattle food resources and in the food industry for development of new products with no lactose content [39]. Cheese whey is a highly polluting product, consisting of 0.7% (w/v) protein, 5% (w/v) lactose, 93% (w/v) water and salts. As a cheap readily available substrate for microbial cell cultivation after the hydrolysis of lactose by β galactosidase this organic waste could be used [40]. Whey proteins (such as α lactalbumin) have excellent functional properties, which can be recovered by ultrafiltration and hydrolyzed to produce many useful pharmaceutical intermediates. The cheese whey/permeate stream can have β galactosidase employed directly to it resulting in high sweetness syrups that can be used as an additive in ice-creams, desserts, etc. [41].
Advantage and Disadvantage
Beta galactosidase is not a good reporter gene for in vivo gene delivery studies. It is difficult to distinguish the exogenous activity as there is too much endogenous activity in most tissues. The amount of DNA expressed when doing non-viral gene delivery is often so low that you cannot overwhelm the endogenous activity and therefore cannot conclusively say the enzyme activity you detected is from your new DNA.
Luciferase is far superior for in vivo work. First, you can be sure that any luciferase activity you detect is real since there is no endogenous luciferase activity in most animals so. Through bioluminescent imaging, luciferase activity can be determined noninvasively allowing for repeated measurements on the same animal. Beta glucosidase can require killing the animal, dissecting the tissues, preparing microscope slides, and then analysis, so doing a time course study will require using many more animals.
The only advantage beta glucosidase has over luciferase is discerning what percentage or precisely what type of cells are expressing the reporter gene. However, GFP or another fluorescent protein would be more appropriate because of the endogenous activity issue described earlier. An insoluble blue dye is formed when X-Gal is hydrolyzed. Evolution of blue color is intensified as more substrate is hydrolyzed, allowing for quick blue/white screening by visual inspection in common applications of cloning and gene expression. B-gal assays are more sensitive to gene expression (the dye precipitates out and retains color very well) using the X-gal substrate, however, X-gal is not quantitative [42].
In contrast, ONPG hydrolysis evolves a soluble yellow dye. This color development is quantitative; but is less sensitive than X-gal because the linear relationship between light absorption (at 420 nm) and substrate concentration is small. Using ONPG as a B-gal substrate allows for quantitative measurement of B-gal activity (and therefore promoter activity) (Note that ONPG itself is not capable of inducing the lac promotor, unlike X-gal).
The choice of reporters depends largely on the question you wish to answer. Luciferase or B-gal with X-gal is best suited for their higher sensitivity if promoter induction is relevant (e.g., the stress-response promoter was activated). X-gal is ideal given that luciferase requires a special camera and optical filter (such as a gel documentation system), because it may be screened by eye. B-gal+ONPG or luciferase will yield quantitative measurements of reporter gene activity, and require at least a spectrophotometer if promoter activity is being assayed.
B-gal is the ideal reporter with the assumption that material costs are to be minimized and ease of experimental protocol to be maximized given these are university labs. On the other hand, luciferase requires: more costly substrate; specialized equipment; careful handling of materials, and more time (as screening necessarily takes longer, therefore lower throughput for a lab demo setting).
Modification Strategy
Biological enzymes unique catalytic activity makes them prime candidates for incorporation into bio-conjugates to perform specific functions. The reactions facilitated by enzymes can be used to enhance synthesis of desired compounds, convert substrates to produce a detectable signal, create active drugs from prodrugs at tumor sites in vivo, and hydrolytically degrade biomolecules to produce defined fragments.
To determine lactose in biological fluids, β-Gal can be used and it is employed in food processing operations, particularly in immobilized form. Often the enzyme is used as a reporter enzyme for monitoring gene activation and transcription.
When conjugated to antibody molecules or streptavidin for use in ELISA systems β-Gal also has good characteristics [43]. Composed of four identical subunits of MW 135,000, each with an independent active site, β-Gal has a molecular weight of 540,000 [44]. With chelated Mg2+ ions required to maintain active site conformation the enzyme has divalent metals as cofactors. Enhanced substrate turnover is caused by the presence of NaCl or dilute solutions (5%) of lowmolecular- weight alcohols (methanol, ethanol, etc.). β-Gal contains numerous sulfhydryl groups and is glycosylated. Commercially available β-gal is usually isolated from E. coli and has a pH optimum at 7 to 7.5.
By contrast, mammalian β-galactosidases usually have a pH optimum within the range of 5.5 to 6; thus, interference from endogenous β-gal during immune-histochemical staining can be avoided. Conjugates formed with antibodies and β-gal can be much bulkier than those associated with alkaline phosphatase or horseradish peroxidase due to the relatively high molecular weight of the enzyme. For this reason, antibody conjugates made with β-gal may have more difficulty penetrating tissue structures during immunehistochemical or immune-cytochemical staining techniques than those made with the other enzymes.
The enzyme remains a minor player in ELISA procedures although numerous research articles have been written describing the preparation and use of antibody conjugates with β-gal. Utilization of this enzyme is at less than 1% of all commercial ELISA products. β-Gal may be conjugated to antibody molecules using the heterobifunctional reagent SMCC. To form a maleimide-activated derivative this cross-linker is reacted first with an antibody through its amine-reactive NHS ester end. This is in contrast with most antibody–enzyme conjugation schemes utilizing SMCC, wherein the enzyme is typically modified first and a sulfhydryl-containing antibody is coupled secondarily.
Nevertheless, conjugations with this enzyme often are done with the antibody being the first targeting molecule and thus enhance detectability, such as dendrimers and polymers since β-gal already contains abundant free sulfhydryl residues that can participate in coupling to a maleimide-activated protein.

View pdf
Download pdf
There are no references
