Journal Name: Journal of Pediatrics and Infants
Article Type: Case Report
Received date: 08 October, 2018
Accepted date: 13 October, 2018
Published date: 18 October, 2018
Citation: Aleman CAP, Vergara RSA, Zarza MCH, Hagood JS, Cortes JLC (2018) Persistent Tachypnea, Hypoxemia and Pulmonary Hypertension Secondary to Pulmonary Neuroendocrine Cell Hyperplasia of Infancy J Pediat Infants. Vol: 1, Issu: 2 (03-06).
Copyright: © 2018 Aleman CAP. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The accurate characterization of childhood interstitial lung disease (ChILD) is critical for prognosis and to determine treatment. ChILD is defined as a heterogeneous group of rare parenchymal lung conditions and has a broad differential diagnosis in early childhood. We present the case of a two-year-old boy with a history of tachypnea and long-standing hypoxemia who developed pulmonary hypertension. The diagnosis of childhood neuroendocrine cell hyperplasia (NEHI) or lung disease associated with neuroendocrine cell hyperplasia was considered based on clinical history and chest computed tomography (CT) findings but required lung biopsy for confirmation. Pulmonary hypertension is rare in NEHI, but can result from longstanding hypoxemia, and does not necessarily have the same negative prognostic significance as it does in other forms of ChILD.
Keywords
Interstitial lung disease, Childhood neuroendocrine cell hyperplasia, Pulmonary hypertension, Computed tomography of the thorax, Lingula and Middle lobe.
Abstract
The accurate characterization of childhood interstitial lung disease (ChILD) is critical for prognosis and to determine treatment. ChILD is defined as a heterogeneous group of rare parenchymal lung conditions and has a broad differential diagnosis in early childhood. We present the case of a two-year-old boy with a history of tachypnea and long-standing hypoxemia who developed pulmonary hypertension. The diagnosis of childhood neuroendocrine cell hyperplasia (NEHI) or lung disease associated with neuroendocrine cell hyperplasia was considered based on clinical history and chest computed tomography (CT) findings but required lung biopsy for confirmation. Pulmonary hypertension is rare in NEHI, but can result from longstanding hypoxemia, and does not necessarily have the same negative prognostic significance as it does in other forms of ChILD.
Keywords
Interstitial lung disease, Childhood neuroendocrine cell hyperplasia, Pulmonary hypertension, Computed tomography of the thorax, Lingula and Middle lobe.
Case Description
A 2-year-old male from Mexico City (altitude 2250 meters) presented with a history of persistent tachypnea and increased work of breathing (50-60 retractions per minute), beginning at age 13 months, as well as persistently low oxygen saturation (SaO2), 85-89 % on room air. There was no significant perinatal history. At 10 months of age he was diagnosed with bronchopneumonia and treated with antibiotics, with inadequate recovery. He subsequently presented with three additional episodes of respiratory distress and wheezing, the last time at 14 months of age of sufficient severity to require hospitalization. From then on, his general condition worsened, and his symptoms (cough, dyspnea and wheezing) were exacerbated by gastroesophageal reflux disease (GERD) which responded to conventional treatment.
His respiratory symptoms persisted requiring re-hospitalization at 19 months of age, when pulmonary hypertension was diagnosed by echocardiography with estimated pulmonary artery pressure values of up to 55 mm Hg.
He was treated with oral steroids, bronchodilators, diuretics, sildenafil and continuous supplemental oxygen up to 0.5 L/min, with continuous measurement of SaO2 to maintain saturation between 94-96 %. Polysomnography was performed to rule out obstructive sleep apnea (OSA). At the end of the treatment, it was brought to room air with SaO2 improvement, decreasing the need for external oxygen.
High resolution chest computed tomography (CT) was performed, demonstrating patchy areas of ground glass attenuation predominantly in the right middle lobe and lingula with areas of air trapping, suggesting small airway involvement and raising the suspicion of neuroendocrine hyperplasia of infancy (NEHI). A open lung biopsy was performed confirming the diagnosis. In the pulmonary parenchyma, prominent congestion with extravasation of erythrocytes into the air and interstitial spaces without hemosiderin deposition was identified. There was no inflammation or significant fibrosis of the interstitium. Some branches of pulmonary arteries were increased in diameter with intima - media hypertrophy, consistent with pulmonary hypertension. Some bronchi and bronchioles with denudated epithelium were noted. Chromogranin staining denoted neuroendocrine cells in the epithelium of the airway (up to 24% of cells). Focal slight accumulation of peribronchial and peribronchiolar lymphocytes was noted. There was minimal submucosal fibrosis of the small airways (constrictive bronchiolitis). Focal alveolar distension was noted, consistent with air trapping. There was no evidence of viral cytopathic changes or granulomas (Figure 1-4).
Figure 1
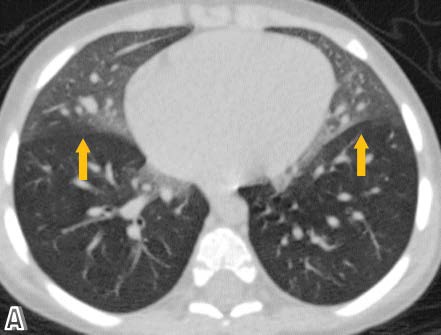
Figure 1: High resolution tomographic images with a pulmonary window in axial.
Figure 2
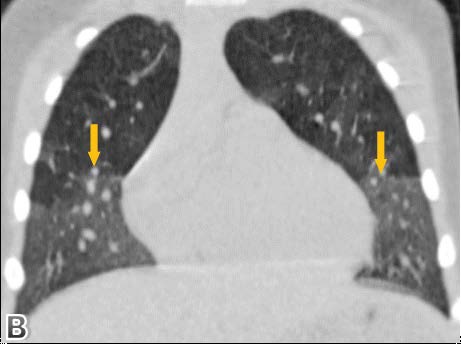
Figure 2: coronal and sagittal planes.
Figure 3
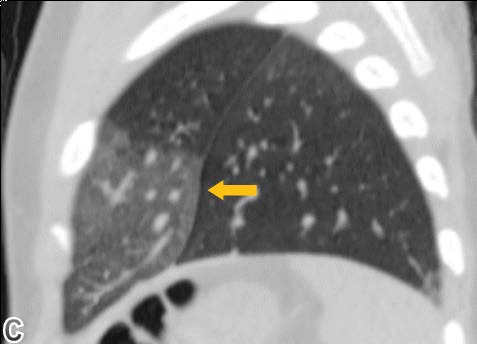
Figure 3: Showing ground glass opacification mainly involving the right middle lobe and left lingular segment.
Figure 4
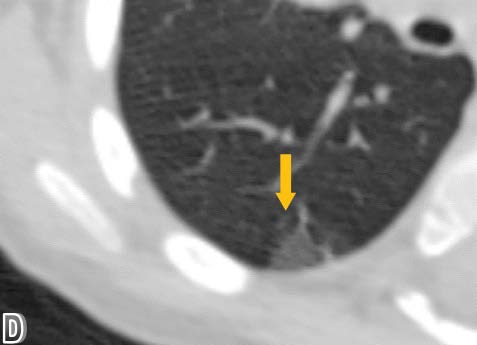
Figure 4: An enlarged axial plane image in the pulmonary window showing a subpleural area of ground-glass opacification.
Discussion
Childhood interstitial lung diseases (ChILD), a subset of diffuse lung diseases, are a diverse group of rare disorders that cause remodeling of the distal airspaces of the lung, resulting in impaired gas exchange. Although they are individually rare, there are many different forms of diffuse lung disease in children, and collectively these disorders are associated with significant morbidity and mortality, as well as with the disproportionate use of medical care resources. In recent years, there have been many significant advances in the field, including genetic discoveries and the development of clinical practice guidelines. The reported prevalence rate of ChILD disorders varies between 0.13 to 16.2 cases per 100,000 children, and these are more common in infants than in older children [1-5].
ChILD syndrome is defined the presence of 3 of the following 4 criteria, after ruling out common disorders such as cystic fibrosis, infection, aspiration and cardiovascular disease:
- Respiratory symptoms (cough, tachypnea or dyspnea of exercise)
- Respiratory signs (tachypnea, crackles, intercostal printing, digital clubbing, difficulty in weight gain)
- Hypoxemia
- Diffuse anomalies in radiography or chest CT
Differentiating among different diagnoses in an infant with ChILD syndrome is important in order to define treatment, monitor clinical course, assess for complications, and define prognosis. The current classification system for interstitial lung diseases in infants includes: 1) diffuse developmental disorders (acinar and alveolar dysplasia), 2) alveolar growth abnormalities, 3) disorders of surfactant dysfunction, and 4) specific conditions of unknown etiology. The fourth subtype includes neuroendocrine cell hyperplasia (NEHI) and pulmonary interstitial glycogenosis [2,6].
NEHI was first described in 2005, presenting with symptoms of ILD in the first year of life, such as persistent tachypnea, retractions, hypoxemia, and crackles. Previously the term persistent tachypnea of infancy had been used to describe this clinical presentation. The only consistent histopathological abnormality was an increase in clear cells in distal airways, consistent with pulmonary neuroendocrine cells (PNEC), confirmed by immunohistochemistry with antibody to bombesin [7,8]. PNEC can also be visualized with antibodies to other neuropeptides such as Chromogranin A, synaptophysin, and CD56 [9].
NEHI commonly presents after a respiratory infection and may be misdiagnosed as recurrent bronchiolitis. Poor growth that requires nutritional supplements and gastroesophageal reflux occur commonly in children with NEHI. The average age at which symptoms present is approximately 4 months [10,11].
The etiology of NEHI is unknown. A small percentage of cases is familial. A genetic basis for a familial form of NEHI associated with a sequence variant in NKX2.1 has been recently described, and PNEC hyperplasia was noted in a familial lung disease associated with surfactant protein C gene mutation. There are no known genetic associations with sporadic cases of NEHI [12-14].
The role of PNECs in the lung is not fully defined; they are granular epithelial cells, often innervated, distributed in the distal conducting airways, often at branch points, making up less than 1% of the epithelial cells in the distal air passages. Sometimes PNECs comprise small clusters called neuroepithelial bodies (NEB). In response to various stimuli, PNECs produce bioactive products (bombesin, serotonin and calcitonin type peptides) that can cause bronchoconstriction, vasomotor activity, epithelial differentiation and alterations of the bronchial smooth muscle. They are present in greater numbers in fetal life, where they are thought to play a role in lung development. They have also been described as increased in several diseases associated with hypoxemia and lung damage such as bronchopulmonary dysplasia, sudden infant death syndrome, pulmonary hypertension, and cystic fibrosis. Quantitative analysis of PNECs using histochemical staining has determined that in NEHI, the number of PNECs and NEBs are significantly increased compared to other lower airway diseases and to normal lungs [12].
A recent study using genetic inactivation of PNECs determined that they are essential for development of inflammatory responses and mucus goblet cell hyperplasia in response to ovalbumin (OVA)-mediated hypersensitivity, confirming their role in sensing foreign substances in the airway and defining a novel immunomodulatory role [15]. The pathophysiologic mechanisms causing excess PNECs in NEHI, and the mechanisms by which their presence is associated with hypoxemia, crackles and increased work of breathing, have yet to be determined.
The diagnostic criteria for NEHI are based on clinical, imaging and histological exams. More common diagnoses should be ruled out first. If available, infant pulmonary function testing usually shows airflow obstruction and air trapping. Chest radiographic findings are variable; they may be normal or demonstrate hyperinflation with variable degrees of peri-hilar opacity. Although definitive diagnosis requires lung biopsy, with the histological finding of an increased number of bombesin antibody-positive neuroendocrine cells to in the bronchioles, high-resolution chest CT seems to be the most reliable non-invasive entity that can suggest the diagnosis of neuroendocrine cell hyperplasia. The findings of multilobar or patchy groundglass opacities that predominantly affect the middle and lingular lobes, as well as the pattern of patchy air trapping, with areas of normal lung are characteristic findings on CT with sensitivity of 83% and a specificity until 100%. However, the characteristic CT pattern should not be considered as pathognomonic of this pathology. More importantly, chest CT can demonstrate other patterns of interstitial lung disease, such as septal thickening or fibrosis, which would suggest other etiologies. Protein surfactant mutations should be sought, and lung biopsies should still be considered for the definitive differential diagnosis [16,17].
Pulmonary hypertension (PH) in childhood has been associated with conditions complicated by chronic hypoxemia, such as interstitial lung diseases. Increase in PNEC are observed in bronchopulmonary dysplasia, where airway and alveolar fibrosis, epithelial metaplasia, and airway and vascular smooth muscle hypertrophy contribute to marked pathophysiologic changes in lung function. Considering the biologic effects of PNEC secretory products, particularly gastrin-releasing peptide on airway epithelial cell and fibroblast proliferation, increase in PNEC secretory products in the regenerating airway epithelium may contribute to the development of the pathologic alterations in lung structure seen in bronchopulmonary dysplasia. Secretion of abnormally large amounts of bronchoactive and vasoactive agents from PNEC (e.g., serotonin, gastrinreleasing peptide) in response to airway hypoxia and hypercapnia may be partially responsible in the genesis of reactive airway disease and pulmonary hypertension seen in this disease [18,19,14].
The prognosis for children with ILD is highly variable, with pulmonary hypertension (PH) being the single greatest clinical predictor of mortality. Infants with NEHI generally have a favorable prognosis, although they may remain symptomatic and require long term oxygen. In this case, and perhaps in other cases of NEHI associated with PH, the presence of PH likely reflected the longstanding hypoxemia and was reversible with oxygen supplementation, and thus did not indicate a worse prognosis. Ongoing research and collaborative efforts are expected to provide insights into the molecular basis of NEHI and other forms of child, and to identify targets for therapeutic intervention, thus allowing the establishment of evidence-based therapies [20,5].
Conclusion
Due to the low incidence and prevalence, nonspecific symptoms, and previous lack of a classification system, ILD in infants has posed diagnostic challenges in the past. However, the establishment of a comprehensive classification system has substantially improved understanding of this challenging category of lung disease. Neuroendocrine cell hyperplasia of infancy (NEHI) should be considered in the differential diagnosis of persistent or recurrent respiratory symptoms in infants in the first years of life, with or without hypoxemia or chest radiograph abnormalities. High resolution chest CT has characteristic findings that can be used as a noninvasive test to support clinical suspicion and also direct the biopsy to the affected areas, but it is critical to rule out more common causes of diffuse lung disease in infants. Pulmonary hypertension is unusual in NEHI and requires more thorough investigation when present. When it occurs secondary to untreated hypoxemia, it may have a better prognosis than when it is present in other childhood ILDs.

View pdf
Download pdf
There is no references
